
一根電極如何「對話」一個失靈的迴路?從帕金森氏症的深部腦刺激談起
把神經疾患從「哪個細胞壞了」升級為「哪條迴路失衡、病理如何沿網路擴散」:基底核直接/間接路徑、DBS 與 β 震盪、連結組擴散方程式與 ATN 生物標記框架。
一根電極如何「對話」一個失靈的迴路?從帕金森氏症的深部腦刺激談起
想像一位帕金森氏症(Parkinson's disease, PD)患者,藥物已經控制不住越來越嚴重的顫抖與動作遲緩。神經外科醫師在他清醒的狀態下,把一根直徑不到 1.3 公釐的電極,精準插進大腦深處一個名叫視丘下核(subthalamic nucleus, STN)的結構——這個結構只有黃豆大小。當醫師打開電流,患者顫抖的手,在幾秒鐘內安靜了下來。
這不是科幻場景,而是已執行數十萬例的深部腦刺激(deep brain stimulation, DBS)手術。但它留下一個迷人的問題:我們明明沒有「補回」任何死掉的多巴胺神經元,為什麼用高頻電流去刺激一個「下游」的核團,就能讓症狀大幅緩解?
入門篇告訴你帕金森氏症源於黑質多巴胺神經元死亡。但「多巴胺不夠」這個分子層次的答案,無法解釋為什麼電刺激 STN 會有效,也無法解釋為什麼同樣是基底核疾病,亨丁頓舞蹈症(Huntington's disease)的症狀卻與帕金森氏症幾乎相反(動作過多而非過少)。要回答這些問題,我們得從「分子」這一層往上爬一層——進入迴路(circuit)的世界。這篇進階文章,邀請你把神經疾患從「哪個細胞壞了」的故事,升級成「哪條迴路失衡了」與「壞掉如何沿著網路擴散」的故事。
基底核的「煞車與油門」:直接與間接路徑模型
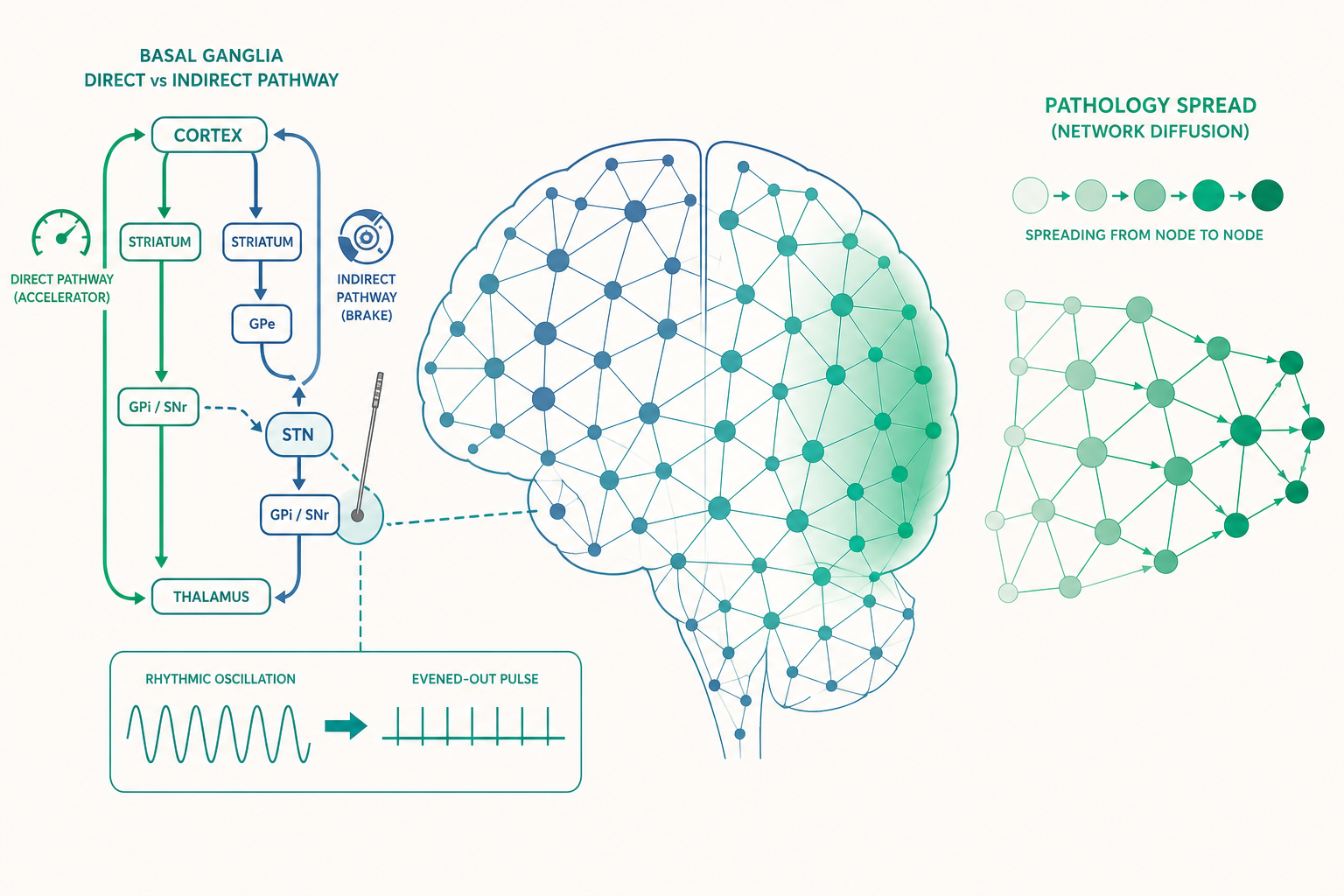
要理解 DBS 的魔力,先要認識基底核(basal ganglia)的經典電路模型:直接路徑(direct pathway)與間接路徑(indirect pathway)。這是 1989 年由 Albin、Young 與 Penney,以及 DeLong 各自提出、後來成為神經學教科書骨幹的模型。
把它想成汽車的「油門與煞車」:
- 直接路徑像油門:大腦皮質下達「想動」的指令後,這條路徑最終會解除對視丘(thalamus)的抑制,讓視丘把動作訊號送回皮質——也就是「放行」動作。
- 間接路徑像煞車:這條路徑繞經外蒼白球(GPe)與視丘下核(STN),最終加強對視丘的抑制,抑制不想要的動作。
關鍵在於:多巴胺對這兩條路徑的作用方向相反。黑質的多巴胺會「踩油門」(活化直接路徑上帶 D1 受體的神經元),同時「鬆煞車」(抑制間接路徑上帶 D2 受體的神經元)。所以正常情況下,多巴胺是一個讓動作「容易發動」的整體調節者。
現在把多巴胺抽掉,看會發生什麼:
- 油門沒人踩(直接路徑活性下降)→ 動作難以發動。
- 煞車沒人鬆(間接路徑過度活躍)→ STN 與內蒼白球(GPi)異常過度放電 → 視丘被過度抑制 → 皮質收不到「放行」訊號。
結果就是動作遲緩(bradykinesia)與僵直。這就是帕金森氏症在「迴路層次」的解釋:不是大腦哪裡「斷線」,而是油門與煞車的平衡被打破,整個迴路被推向「過度抑制」這一端。
而亨丁頓舞蹈症恰好相反:它的病理是紋狀體(striatum)裡間接路徑的神經元優先死亡——煞車系統壞了,於是動作關不住,出現不自主的舞蹈樣動作(chorea)。同一個迴路、相反的失衡方向、相反的症狀。一旦你掌握這個模型,許多看似零散的運動障礙疾病就被同一張地圖串了起來。
DBS 為什麼有效?「資訊損毀」假說
回到開頭的謎題。既然帕金森氏症時 STN 是「過度放電」的,那最直覺的想法是:用電去抑制它不就好了?早期確實有人這樣猜。但事情沒這麼簡單——高頻 DBS(典型為 130–185 赫茲)施加的明明是刺激,效果卻像把這個過度活躍的核團「關掉」。
目前主流的解釋已經從「DBS 單純抑制神經元」轉向更精緻的「資訊損毀(information lesioning)」假說:高頻規律電刺激會用一個人工的、固定節律的訊號,覆蓋並打亂原本病態的放電模式。
這裡有一個進階且關鍵的觀念:帕金森氏症的問題可能不只是「放電太多」,更是「放電太有規律、太同步」。健康大腦的基底核放電是去同步的、富含訊息的;而在多巴胺枯竭時,STN 與 GPi 出現異常的β 頻段(約 13–30 赫茲)同步震盪(beta oscillations)。許多研究發現,這種病態 β 震盪的強度,與動作遲緩和僵直的嚴重度高度相關——它彷彿是大腦卡在「不要動」狀態的電生理簽名。
於是 DBS 的作用可以重新理解為:用 130 赫茲的規律脈衝去淹沒那個 20 赫茲的病態合唱,讓下游結構不再被錯誤訊號綁架。換句話說,DBS 治療的不是「多巴胺濃度」,而是「迴路的動態與通訊品質」。這是一個從分子治療轉向動態系統治療的範式轉移,也是計算神經科學介入臨床最漂亮的例子之一。
看一個例子:閉迴路 DBS 與「按需供電」
傳統 DBS 是「開迴路(open-loop)」的——電流 24 小時不間斷輸出,不管你此刻是在顫抖還是在睡覺。這既耗電(電池要開刀更換),又可能在不需要時造成副作用(如言語含糊、平衡變差)。
那能不能讓刺激「按需供電」?這就是近年最熱門的閉迴路/適應性 DBS(adaptive DBS, aDBS)。它的邏輯非常工程化,跟你在控制系統課學的回饋控制一模一樣:
- 感測(sense):植入的電極不只放電,也讀取局部場電位(local field potential, LFP),即時偵測那個病態 β 震盪的強度。
- 判斷(decide):一個內建演算法判斷——β 功率超過閾值(代表症狀正在惡化)了嗎?
- 致動(actuate):只有在 β 超標時才送出刺激;症狀緩解、β 下降,就把刺激調低或關掉。
2023–2024 年間,多項臨床試驗已顯示 aDBS 在控制症狀的同時能減少總刺激量與副作用,相關技術也陸續取得監管核准。請注意這個架構的本質:它把一個生物標記(β 震盪)當成回饋訊號,閉合了「感測—運算—致動」的迴路。這正是 AI/控制理論與神經醫學的交會點——大腦不再只是被「治療」的對象,而是一個被持續監測、即時調節的動態系統。
如果你對 Uedu Fit 把 HRV、睡眠當成生理回饋訊號的思維有印象,會發現 aDBS 是同一個哲學的極致版本:用即時生理訊號去閉合一個調節迴路,只是這裡的訊號直接來自大腦深處、致動器是電極而非提醒通知。
從「點」到「網」:神經退化是一種連結組疾病
換到退化性疾病這一側。入門篇提過 tau、α-突觸核蛋白可能像普利昂(prion)一樣傳播。進階篇要把這個想法推到一個更有力的框架:神經退化疾病本質上是「網路疾病(network disorders)」,而非「區域疾病」。
過去我們習慣問:「阿茲海默症先攻擊哪個腦區?」答案是內嗅皮質與海馬迴。但更深刻的問題是:為什麼是這些區域、又為什麼病理會沿著特定路線蔓延?
近十年的網路退化假說(network degeneration hypothesis)給了一個優雅的答案:致病蛋白(tau、α-synuclein、TDP-43 等)不是隨機擴散,而是沿著神經元之間的軸突連結,從一個區域「跳」到與它解剖相連的下一個區域。換句話說,病理擴散的地圖,幾乎就是大腦連結組(connectome)的地圖。
這個假說有幾個強而有力的證據與推論:
- 不同失智症對應不同的腦網路。阿茲海默症的萎縮模式高度吻合所謂的「預設模式網路(default mode network, DMN)」;而額顳葉失智(FTD)的不同亞型,則分別對應語言網路或社會情緒網路。失智症在某種意義上是「特定大腦網路的選擇性崩解」。
- 連結越密、越中樞的區域,越脆弱。大腦裡有些高度連結的「樞紐(hub)」區域,承擔最多的訊息轉運。它們代謝負擔重、蛋白質周轉壓力大,反而成為退化的高風險區——這個現象被形容為「樞紐的代價」。
- 可以用數學模型「預測」萎縮的擴散。研究者把致病蛋白的擴散寫成在連結組網路上的擴散方程式(network diffusion model),給定一個起始點,就能模擬未來幾年萎縮會往哪裡蔓延,並與真實病人的縱貫影像比對。這把神經退化從一個描述性的病理學,變成一個可計算、可預測的動力系統問題。
動手試試:把擴散當成圖論問題
請你先別往下看,試著用你學過的數學重新表述這件事。如果把每個腦區看成一個「節點」、把白質纖維束看成「邊」,整個大腦就是一張圖(graph)。致病蛋白的擴散,能不能寫成這張圖上的方程式?
如果你想到了類似「熱在金屬網格上擴散」的圖像,你已經抓到精髓了。網路擴散模型的核心,正是把蛋白病理的濃度向量 $x$ 隨時間的變化,寫成:
$$\frac{dx}{dt} = -\beta L x$$
其中 $L$ 是這張腦網路的圖拉普拉斯矩陣(graph Laplacian)——它編碼了「哪個節點跟哪個節點相連、連得多強」,$\beta$ 是擴散速率。這個方程式說的是:每個節點上的病理,會依照它與鄰居的連結強度,往濃度低的鄰居流動。
這個練習的意義在於:你會發現同一個圖拉普拉斯工具,既出現在物理的熱傳導、又出現在你可能在資料科學課用過的圖卷積網路(GCN)、現在又出現在預測阿茲海默症萎縮的臨床模型裡。當神經科學被翻譯成圖論語言,它就和你在數學、物理、AI 學到的工具接通了——這正是「優神經科學」想讓你體會的跨域力量。
生物標記與疾病分期:把模糊的「失智」變成可量化的 ATN 框架
進階臨床神經科學還有一個你必須認識的轉變:診斷邏輯的重構。過去阿茲海默症的「確診」幾乎要等到病人過世後解剖看到斑塊與纏結。但這對活著的病人毫無幫助,也讓臨床試驗難以挑對受試者。
於是學界提出了生物標記導向的框架,其中最有影響力的是 ATN 分類系統(由 Jack 等人於 2018 年提出):
- A(Amyloid):類澱粉蛋白病理——可用腦脊髓液檢測或類澱粉 PET 影像評估。
- T(Tau):tau 病理——可用 tau PET 或腦脊髓液 phospho-tau 評估。
- N(Neurodegeneration):神經退化/神經元損傷——可用結構性 MRI 萎縮或 FDG-PET 評估。
這個框架的精妙之處,在於它把疾病從「症狀定義」改成「生物學定義」。一個人可能 A 陽性、T 陰性、N 陰性——代表病理已經啟動,但還沒造成可見的神經退化或症狀。這開啟了「臨床前阿茲海默症(preclinical AD)」這個概念:在記憶出問題的十幾二十年前,大腦裡的分子戰爭可能早已開打。
為什麼這對治療是顛覆性的?因為它呼應了入門篇那個殘酷事實——神經元幾乎不再生。如果等到症狀出現才治療,大量神經元早已死亡,任何藥物都只是亡羊補牢。生物標記讓我們有機會在神經元還活著時就出手,這也是 2023 年起以 lecanemab、donanemab 為代表的抗類澱粉抗體藥物,把臨床試驗對象前移到「早期、生物標記確診」族群的根本原因。
值得平衡指出:這些新藥雖在統計上顯著減緩了認知退化速度,但臨床效益幅度有限,且伴隨腦水腫與微出血(合稱 ARIA)的風險。學界對「值不值得、對誰值得」仍在激辯。這再次提醒我們——入門篇就強調過——類澱粉假說至今仍是開放議題,而非蓋棺論定的真理。進階學習者的素養,正在於能同時握住「這是重要進展」與「這遠非終點」這兩個判斷。
重點回顧
- 從分子到迴路是關鍵升級:帕金森氏症在迴路層次是基底核「直接(油門)/間接(煞車)路徑」失衡、被推向過度抑制;亨丁頓舞蹈症則是煞車先壞,呈現相反症狀。
- DBS 治療的是「動態」而非「濃度」:高頻刺激以「資訊損毀」方式淹沒病態的 β 同步震盪,閉迴路 aDBS 更把 β 當回饋訊號,實現「感測—運算—致動」的按需刺激。
- 神經退化是網路疾病:致病蛋白沿連結組擴散,不同失智症對應不同腦網路,高連結的樞紐區域最脆弱(樞紐的代價)。
- 擴散可被數學化:用圖拉普拉斯矩陣寫成網路擴散方程式,能預測萎縮蔓延路徑,把臨床問題接上圖論與 AI 工具。
- ATN 生物標記框架把疾病重新定義為生物學狀態,揭示「臨床前期」的存在,使治療有機會在神經元死亡前介入——但新藥效益與風險仍在權衡中。
深入探討(研究所視角)
震盪即計算:β 與 γ 不只是雜訊。進階神經科學越來越把神經震盪(neural oscillations)視為大腦協調訊息的「載波」,而非副產品。帕金森氏症的病態 β 震盪、癲癇的異常同步、甚至阿茲海默症中 γ 震盪(約 40 赫茲)的減弱,都暗示「時間結構的失調」本身就是一種病理。一個極具想像力的前沿是「γ 夾帶(gamma entrainment)」研究——讓動物(與初步的人體試驗)暴露在 40 赫茲的閃光或聲音下,觀察是否能驅動皮質 γ 震盪、進而影響類澱粉清除與小膠細胞活性。這條線索把「動態系統」「免疫」「治療」綁在一起,也提醒我們:大腦的疾病,可能藏在「頻率」而不只是「結構」裡。研究所層級的提問會是——病態同步究竟是病因、代償,還是單純的標記?因果方向至今未定。
連結優生物:選擇性脆弱(selective vulnerability)的分子根源。為什麼是黑質多巴胺神經元、而不是隔壁的神經元,在帕金森氏症中優先死亡?一個有力的解釋指向這類神經元的生理特性本身就高耗能、高風險:它們有極度龐大且分支繁複的軸突網路、依賴 L 型鈣通道維持自發放電(造成持續的鈣負擔)、且粒線體承受巨大氧化壓力。換句話說,「選擇性脆弱」可能是這些細胞為了執行特殊功能所付出的代謝代價。這把疾病問題拉回到細胞能量學與粒線體生物學——想深入的學習者,可順著「粒線體品質管制(mitophagy)」與 PINK1/Parkin 這條家族性帕金森氏症基因線索鑽研下去。
連結優心理與計算精神醫學:把基底核模型用到「決策」。基底核不只管動作,也管動作的選擇與獎賞學習。多巴胺在計算神經科學裡有一個著名身分——獎賞預測誤差(reward prediction error)訊號,這正是強化學習(reinforcement learning)的核心。於是帕金森氏症患者的多巴胺缺損,不只造成動作遲緩,也被發現會影響從回饋中學習的方式(例如更難從正向回饋中學習,卻較能從懲罰中學習)。這條線索開啟了計算精神醫學(computational psychiatry)這個新興領域:用形式化的學習與決策模型,去刻畫帕金森氏症、憂鬱、成癮等疾病在「價值計算」上的偏差。對修過機器學習的你,這意味著 Bellman 方程式與時序差分學習,竟能成為理解一個神經疾病的工具。
連結 AI:從「預測影像」到「個人化的疾病軌跡」。入門篇談了 AI 做早期偵測;進階視角更想強調疾病進程模型(disease progression modeling)。真實病人的退化速度差異巨大,而像 SuStaIn 這類機器學習模型,能從一群病人的橫斷面資料中,同時推斷出疾病的「分期」與「亞型」——也就是不假設所有人走同一條路,而是讓資料自己浮現出多條平行的退化軌跡。這對臨床試驗極為重要:若能事先把病人歸到正確的亞型與分期,就能挑出最可能對某藥物有反應的族群,大幅提升試驗成功率。當神經疾患的最大難題之一是「異質性(heterogeneity)」——同一個病名底下其實是好幾種不同的生物學過程——AI 最擅長的「從雜亂中找出隱藏結構」正好對症下藥。
一個值得帶走的整合視角。入門篇教你在「症狀—解剖—分子—治療」之間穿梭;進階篇想再加兩個維度:動態(dynamics)與網路(network)。看到帕金森氏症,你不只想到多巴胺,還想到基底核迴路的失衡、β 震盪的病態同步、以及 DBS 如何用電流重塑這個動態;看到阿茲海默症,你不只想到斑塊,還想到 tau 如何沿連結組擴散、如何用圖拉普拉斯方程式預測它的下一步、如何用 ATN 框架在症狀出現前攔截它。當你能把「壞掉的細胞」「失衡的迴路」「擴散的網路」「可計算的動力學」這四層串成一條鏈,你對神經疾患的理解,就從一張病理圖譜,升級成一個關於大腦作為動態網路系統,如何運作、如何崩解、又如何可能被即時調節與守護的完整框架。