
為什麼你可以憋氣,卻憋不了太久?
從壓力差驅動的呼吸力學、肺泡氣體交換、血紅素運送,到腦幹以二氧化碳與 pH 為主軸的呼吸調控。
為什麼你可以憋氣,卻憋不了太久?
試著現在深吸一口氣,然後憋住。一開始很輕鬆,但十幾秒後,胸口開始發緊,一股「非呼吸不可」的衝動湧上來,最終你不得不大口換氣。許多人以為這股衝動來自「身體缺氧」,但真相恰恰相反:驅使你重新呼吸的主因,並不是血液裡氧氣(O₂)變少,而是二氧化碳(CO₂)累積、血液變酸。這個看似違反直覺的事實,正是理解整個呼吸系統的鑰匙——呼吸不只是「把氧氣吸進來」,更是一套精密的化學調控系統,分分秒秒守護著體內的酸鹼平衡。
這篇文章將帶你從一次呼吸的物理過程出發,逐步深入到肺泡裡的氣體交換、血液如何運送氣體,以及大腦如何在你毫無察覺的情況下調控每一次吸吐。
一次呼吸的力學:壓力差驅動氣流
呼吸的本質是「氣體順著壓力差流動」。空氣會從壓力高的地方流向壓力低的地方,而我們的身體所做的,就是主動改變胸腔內的壓力。
吸氣時,最重要的肌肉是橫膈膜(diaphragm)——一片位於胸腔與腹腔之間的圓頂狀肌肉。當橫膈膜收縮,它會向下壓平,同時肋間外肌(external intercostal muscles)收縮把肋骨往上、往外提起。胸腔容積因此增大,依據波以耳定律(Boyle's law,氣體體積與壓力成反比),肺內壓力下降到略低於大氣壓,空氣便自然流入肺部。
吐氣在平靜狀態下則是被動的:橫膈膜與肋間肌放鬆,肺與胸壁因本身的彈性回縮(elastic recoil)而縮小,肺內壓力升高,空氣被擠出。只有在用力吐氣(如咳嗽、吹氣球、劇烈運動)時,腹肌與肋間內肌才會主動參與。
這裡有一個關鍵卻常被忽略的結構:胸膜腔(pleural cavity)。肺臟外覆兩層胸膜,兩層之間是一個密閉、帶有負壓的薄液體層。這個負壓像「吸盤」一樣,讓肺臟緊貼著胸壁,隨胸腔擴張而被動撐開。一旦這個密閉空間被破壞——例如外傷或肺泡破裂導致空氣進入胸膜腔——就會發生氣胸(pneumothorax),肺臟失去牽引而塌陷,這也是為什麼胸部穿刺傷會危及生命。
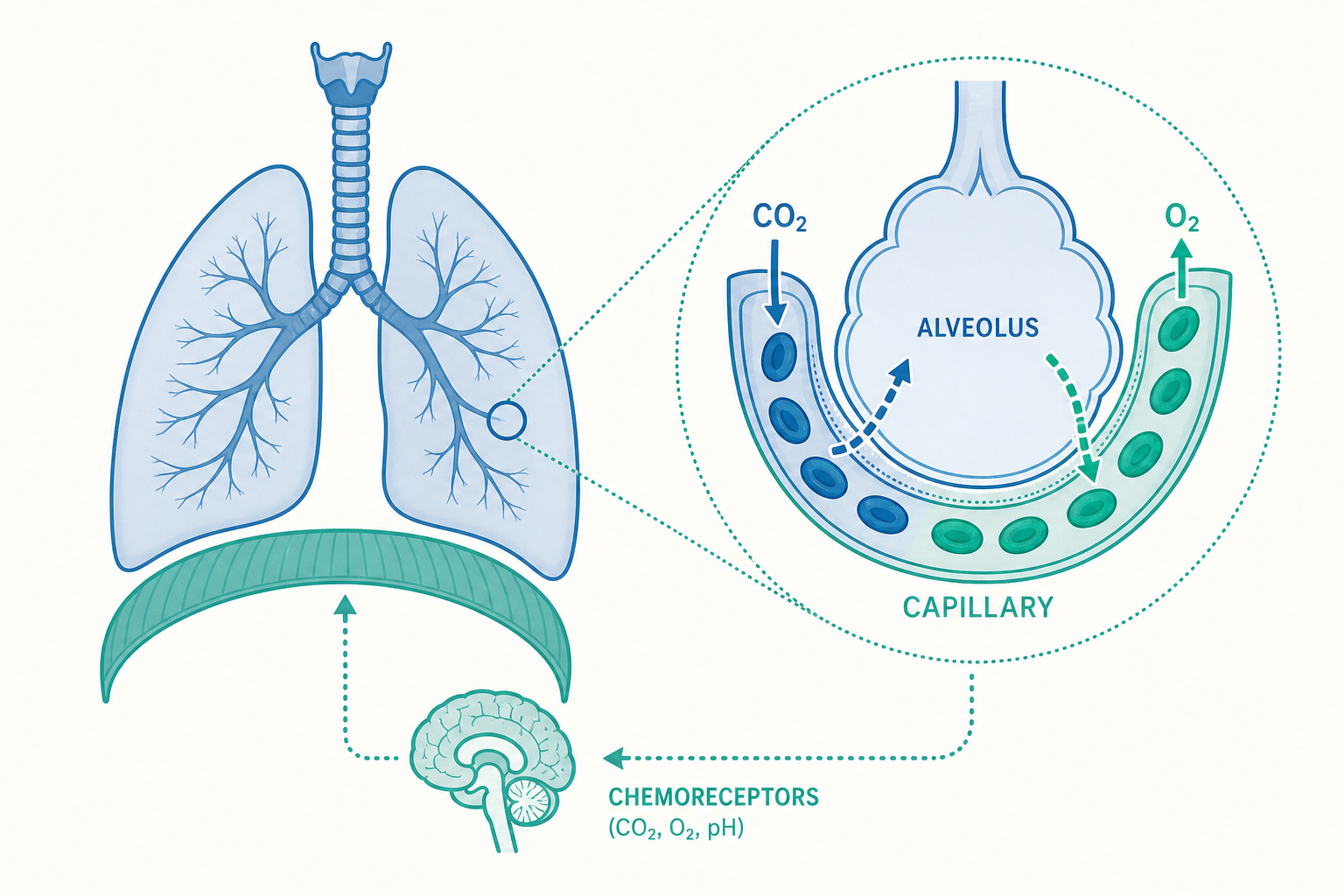
氣體交換的舞台:肺泡與呼吸膜
吸進來的空氣最終要抵達肺泡(alveoli)——這才是真正進行氣體交換的場所。人體約有三億個肺泡,全部攤開的總表面積約 70 平方公尺,接近半個羽毛球場。這個驚人的表面積,正是高效率氣體交換的物理基礎。
氣體交換發生在「呼吸膜」(respiratory membrane)上,它薄到只有約 0.5 微米,由三層構成:肺泡上皮細胞、共用的基底膜、以及微血管內皮細胞。氣體透過簡單擴散(simple diffusion)穿過這層薄膜,遵循費克擴散定律(Fick's law of diffusion):擴散速率與表面積、壓力差成正比,與膜厚度成反比。
這也解釋了多種肺部疾病的本質: - 肺氣腫(emphysema):肺泡壁被破壞、互相融合成大囊泡,表面積大幅減少,氣體交換效率下降。 - 肺纖維化(pulmonary fibrosis):呼吸膜增厚、變硬,擴散距離拉長,氧氣難以通過。 - 肺水腫(pulmonary edema):液體積在肺泡與微血管之間,等於在擴散路徑上多塞了一層水。
氣體擴散的方向由分壓差(partial pressure gradient)決定。肺泡內氧分壓約 104 mmHg,而流經肺部的靜脈血氧分壓只有約 40 mmHg,氧氣便從肺泡擴散進血液;二氧化碳則相反,從分壓較高的血液(約 45 mmHg)擴散進肺泡(約 40 mmHg)排出體外。
血液如何運送氣體:血紅素的精巧設計
氧氣在水中(血漿)的溶解度極低,光靠血漿溶解根本不足以供應全身。解決方案是血紅素(hemoglobin, Hb)——紅血球內的含鐵蛋白,約 98.5% 的氧氣是與血紅素結合運送的,只有約 1.5% 溶於血漿。
每個血紅素分子有四個血基質(heme)位置,可結合四個氧分子。血紅素與氧的結合呈現一條 S 形(sigmoid)的氧合血紅素解離曲線(oxygen-hemoglobin dissociation curve)。這條曲線的形狀來自「協同效應」(cooperativity):當第一個氧分子結合後,會誘導血紅素構型改變,使後續氧分子更容易結合。
S 形曲線在生理上極為巧妙: - 在肺部(高氧分壓區):曲線進入平坦的高原段。這意味著即使你在高海拔、氧分壓略降,血紅素仍能維持接近飽和,氧氣攜帶量不會驟降。 - 在組織(低氧分壓區):曲線位於陡峭段。組織氧分壓只要稍微下降,血紅素就會大量釋放氧氣,正好滿足代謝旺盛組織的需求。
曲線還會「左右移動」來精細調節: - 波耳效應(Bohr effect):當組織代謝旺盛、CO₂ 增多、pH 下降、溫度上升時,曲線右移,血紅素對氧的親和力降低,更願意把氧氣交給最需要的組織。 - 2,3-雙磷酸甘油酸(2,3-BPG):在慢性缺氧(如高海拔適應)時增加,同樣使曲線右移,促進組織供氧。
二氧化碳的運送則更有層次:約 70% 以碳酸氫根離子(bicarbonate, HCO₃⁻)的形式溶於血漿,約 23% 與血紅素結合成碳醯胺基血紅素(carbaminohemoglobin),其餘約 7% 直接溶解。其中碳酸氫根的形成由紅血球內的碳酸酐酶(carbonic anhydrase)催化:CO₂ + H₂O ⇌ H₂CO₃ ⇌ H⁺ + HCO₃⁻。這個反應正是把「呼吸」與「酸鹼平衡」連結起來的核心化學式。
看一個例子:一氧化碳中毒為什麼致命?
理解了血紅素,就能理解一氧化碳(CO)中毒的危險。一氧化碳與血紅素的結合力約是氧氣的 200 至 250 倍。當你吸入含 CO 的氣體(如不完全燃燒的瓦斯、汽車廢氣),CO 會搶占血紅素上的結合位置,形成碳氧血紅素(carboxyhemoglobin),讓血紅素無法攜帶氧氣。
更陰險的是,CO 不只「搶位置」,還會使剩下的血紅素解離曲線左移,使已結合的氧氣更不願意釋放給組織——雙重打擊。患者的動脈血氧分壓(PaO₂,溶解態)可能看起來正常,因為溶解在血漿裡的氧氣沒受影響,一般指尖血氧機(脈搏血氧儀)甚至會誤判為正常,但實際上組織正在嚴重缺氧。這也是為什麼 CO 中毒的治療要用高濃度純氧、甚至高壓氧,藉由大量提高溶解態氧氣與競爭結合位置,把 CO 從血紅素上逐出。
(以上為機制說明與衛教,非個人醫療建議;若疑似中毒請立即就醫並撥打急救電話。)
呼吸的自動駕駛:腦幹如何調控
你不需要「記得」呼吸,因為呼吸的節律由腦幹(brainstem)自動產生。延腦(medulla oblongata)中的呼吸節律中樞會發出規律的神經衝動,控制吸氣與吐氣肌肉;橋腦(pons)則協助讓呼吸更平順。
而真正決定「呼吸要多快多深」的,是化學感受器(chemoreceptors)的回饋:
- 中樞化學感受器(central chemoreceptors):位於延腦,是調控呼吸的主力。它們其實偵測的不是 CO₂ 本身,而是腦脊髓液(CSF)的 pH 值。當血液 CO₂ 升高,CO₂ 能輕易穿過血腦障壁進入腦脊髓液,與水反應生成 H⁺,使 pH 下降,化學感受器立刻偵測到並加快、加深呼吸,把多餘的 CO₂ 排出。
- 周邊化學感受器(peripheral chemoreceptors):位於頸動脈體(carotid body)與主動脈體(aortic body),主要偵測動脈血氧分壓。但它們只有在 PaO₂ 大幅降到約 60 mmHg 以下時才會強烈反應,屬於「緊急備援系統」。
這正是文章開頭謎題的答案:日常呼吸驅動主要來自 CO₂/pH 的變化,而非缺氧。你憋氣時,是累積的 CO₂ 讓中樞化學感受器發出強烈警報,逼你呼吸。
動手試試:過度換氣的小實驗(請謹慎)
如果你刻意做幾次快速深呼吸(過度換氣,hyperventilation),可能會感到頭暈、手指發麻。為什麼?因為你把過多 CO₂ 排出體外,血液 CO₂ 下降、pH 上升(呼吸性鹼中毒,respiratory alkalosis),導致腦血管收縮、血鈣的游離形式改變而出現手腳麻刺感。
這個現象反過來驗證了一件事:CO₂ 在血液中扮演的角色遠不只是「廢氣」,它是維持酸鹼恆定的關鍵旋鈕。也因此,過度換氣造成的不適,傳統上會建議用「對著紙袋呼吸」重新吸回部分 CO₂——但此法有風險(不適合本身就缺氧者),現代臨床更傾向引導患者放慢呼吸節律。請勿勉強進行此實驗,尤其有心血管或癲癇病史者應避免。
從鼻腔到肺泡:常被低估的氣道功能
氣體交換固然是肺泡的工作,但通往肺泡的「導氣部」(conducting zone)絕非只是管道。從鼻腔、咽、喉、氣管到逐層分支的支氣管,它們負責:
- 加溫與加濕:空氣抵達肺泡前已被調節到接近體溫、近乎飽和濕度,保護脆弱的呼吸膜。
- 過濾與清除:鼻毛攔截大顆粒;氣道內覆有黏液與纖毛,形成「黏液纖毛電扶梯」(mucociliary escalator),把吸入的灰塵、病原往上推送排出。吸菸會癱瘓纖毛,這也是吸菸者容易反覆感染與慢性咳嗽的原因之一。
- 解剖無效腔(anatomical dead space):導氣部約 150 毫升的空間不參與氣體交換。這解釋了為什麼「淺而快」的呼吸效率較差——每次吸氣有相當比例只填滿了無效腔,沒抵達肺泡。深而慢的呼吸反而能讓更多新鮮空氣真正進入交換區。
重點回顧
- 呼吸是壓力差驅動的力學過程:橫膈膜與肋間肌收縮使胸腔擴張、肺內壓下降,空氣流入;平靜吐氣靠肺的彈性回縮,是被動的。
- 氣體交換靠擴散,遵循費克定律:薄而廣的呼吸膜(約 0.5 微米厚、70 平方公尺)讓氧氣與二氧化碳順著分壓差快速交換;肺氣腫、肺纖維化、肺水腫各自破壞此過程的不同環節。
- 氧氣主要由血紅素運送:S 形解離曲線兼顧「肺部高效裝載」與「組織充分卸載」;波耳效應讓代謝旺盛處獲得更多氧。
- 二氧化碳多以碳酸氫根運送,碳酸酐酶把呼吸與酸鹼平衡緊密連結。
- 日常呼吸驅動來自 CO₂/pH:中樞化學感受器偵測腦脊髓液 pH 為主力,缺氧只是周邊化學感受器的緊急備援——這正是「憋不住氣」的真正原因。
深入探討(研究所視角)
通氣/灌流匹配(V/Q matching)與低氧性肺血管收縮
真實的肺臟並非每個肺泡都得到等量的通氣(ventilation, V)與血流灌流(perfusion, Q)。理想的氣體交換要求 V/Q 比值接近 1,但受重力影響,直立時肺尖通氣相對過多(V/Q 偏高,趨近無效腔效應),肺底則灌流相對過多(V/Q 偏低,趨近分流效應)。肺臟有一個獨特的調節機制——低氧性肺血管收縮(hypoxic pulmonary vasoconstriction, HPV):當某區肺泡氧分壓下降,該區的肺小動脈會主動「收縮」(與全身血管遇缺氧便舒張的反應恰好相反),把血流導向通氣較好的肺區,藉此優化整體 V/Q 匹配。這個機制在局部肺炎時是有益的代償,但在慢性、全肺性缺氧(如高海拔、慢性阻塞性肺病)時,普遍性的 HPV 會推高肺動脈壓力,長期導致肺動脈高壓與右心衰竭(肺源性心臟病,cor pulmonale)。
呼吸節律的細胞起源:前包欽格複合體
腦幹如何「自發」產生節律,曾是長年謎題。1991 年研究者在延腦腹外側發現了前包欽格複合體(pre-Bötzinger complex),被視為吸氣節律的核心起搏器。其神經元具備內在的節律放電特性,透過興奮性突觸網絡同步化,產生穩定的吸氣節奏。這項發現把呼吸控制從「黑盒子」推向可解析的神經迴路層次,也為中樞性睡眠呼吸中止、嬰兒猝死症等病理提供了研究切入點。
酸鹼生理的整合視角:呼吸與腎臟的代償雙人舞
呼吸系統與腎臟共同維持血液 pH(正常約 7.35–7.45),可用 Henderson-Hasselbalch 方程式描述:pH ∝ [HCO₃⁻] / (PaCO₂)。呼吸系統透過調整 PaCO₂ 進行「快速」代償(數分鐘內),腎臟則透過調節 HCO₃⁻ 的再吸收與排泄進行「緩慢」代償(數小時至數天)。例如代謝性酸中毒(如糖尿病酮酸血症)時,會出現深快的庫斯莫呼吸(Kussmaul breathing),藉過度換氣排出 CO₂ 來代償;慢性呼吸性酸中毒(如慢性阻塞性肺病)時,腎臟則會增加 HCO₃⁻ 保留來緩衝。理解這套雙系統代償,是臨床判讀動脈血液氣體分析(ABG)的核心,也是呼吸生理跨越到腎臟生理、急重症醫學的橋樑。
跨領域連結:從演化到工程
呼吸系統的設計也呼應了演化與物理工程的取捨。哺乳類採「潮汐式」呼吸(空氣進出同一管道),存在無效腔與殘氣的代價;而鳥類演化出「單向氣流」的氣囊系統,氣體交換效率更高,支撐了高耗氧的飛行與高海拔遷徙。這類比較生理學的觀點,正啟發著生醫工程領域——從人工肺(ECMO,葉克膜)的膜式氧合器設計,到仿生氣體交換裝置,都在向自然界這套運作了數億年的擴散系統取經。對有志於生理學、生醫工程或急重症醫學的學習者而言,呼吸系統是一個絕佳的起點:它同時融合了力學、化學、神經調控與演化生物學,是「整合生理學」精神的最佳縮影。