
血壓正常,病人卻在死亡邊緣:休克的隱形時鐘
從 DO₂/VO₂、乳酸與四型休克機制,理解為何血壓「正常」也可能正在崩潰,以及現代復甦的流體反應性與損害控制思維。
血壓正常,病人卻在死亡邊緣:休克的隱形時鐘
急診室送來一位車禍後的年輕女性,意識清楚、血壓 118/76 mmHg,看起來「還好」。但她的心跳 122 次/分、皮膚濕冷、抽血乳酸(lactate)高達 5.8 mmol/L。三十分鐘後,她的血壓突然崩落到 70/40 mmHg。為什麼一個「血壓正常」的病人,會在短時間內急轉直下?
入門篇談的是「在現場辨識心跳停止、做 CPR 與去顫」——那是循環已經完全崩潰的終局。但臨床上更常見、也更需要功力的,是病人「還沒倒下、但正在倒下」的那段灰色地帶。這就是休克(shock)的世界:組織的氧氣供應已經跟不上需求,細胞層級的代謝危機正在進行,而傳統的血壓計卻可能毫無警示。本篇要把鏡頭從「停止後的搶救」拉回到「停止前的血流動力學」,看懂急重症醫師如何在指標還「正常」時就讀出危機。
請注意:本文為醫學知識讀本,目的在於建立機制理解,並非個人化的醫療建議。實際遇到緊急狀況,請立即撥打 119 並依受過訓練的醫護人員指示行動。
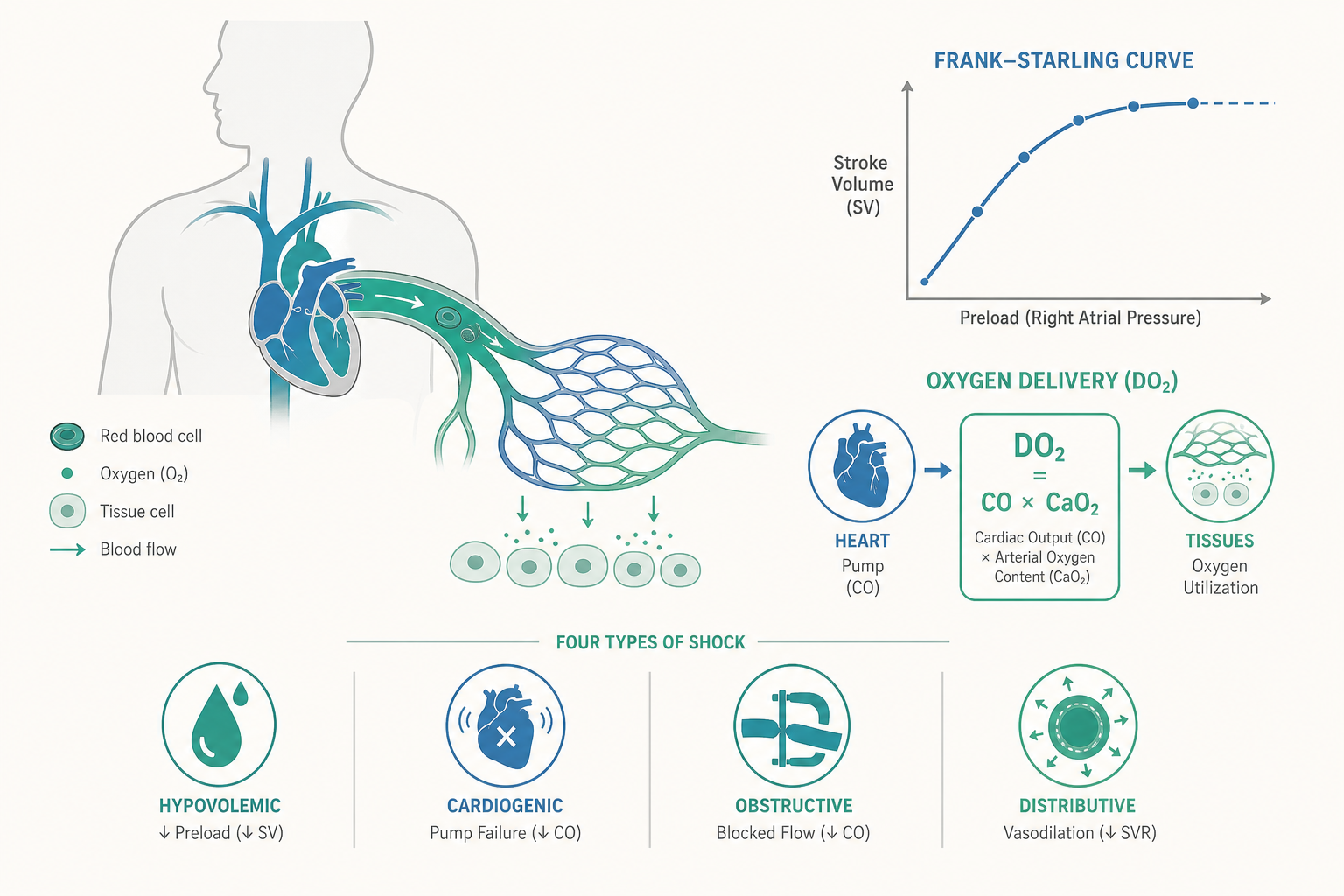
重新定義休克:不是「血壓低」,而是「細胞缺氧」
最常見的迷思,是把休克等同於低血壓(hypotension)。這個直覺害人不淺。休克的本質是組織的氧氣供應不足以維持細胞的有氧代謝,低血壓只是它「可能」的表現之一,而且往往是晚期才出現的表現。
關鍵在於人體強大的代償機制。當有效循環容量下降,交感神經被活化、兒茶酚胺(catecholamine)大量釋放,會讓周邊血管收縮、心跳加速,把血壓「撐」在正常範圍。年輕、健康的病人代償能力特別強,可能失血達總血量的 30% 仍維持收縮壓在正常值——這就是開頭那位車禍病人「血壓正常」的真相。她不是沒事,而是身體正在用盡全力代償。一旦代償機制耗竭,血壓會在沒有太多預警下「斷崖式」下墜。
這帶出一個臨床鐵則:等到血壓掉下來才認定休克,往往已經錯過了黃金處置窗。代償期的休克(compensated shock)才是真正考驗辨識能力的階段。我們需要的,是能在血壓崩潰之前就洩漏訊息的指標。
氧氣供需的數學:DO₂ 與 VO₂
要量化「氧氣供應跟不上需求」,急重症醫學用兩個核心概念:氧氣輸送(oxygen delivery, DO₂)與氧氣消耗(oxygen consumption, VO₂)。
氧氣輸送 DO₂ 描述每分鐘能送到組織多少氧氣,可拆解為:
DO₂ ≈ 心輸出量(CO)× 動脈血氧含量(CaO₂)
而動脈血氧含量主要由血紅素濃度與其飽和度決定:
CaO₂ ≈ 1.34 × 血紅素(Hb)× SaO₂ +(少量溶解氧)
把這兩條式子合起來看,就能理解為什麼休克的成因可以分門別類:心輸出量不足(幫浦或容量問題)、血紅素太低(出血)、或飽和度太低(呼吸衰竭),任何一項崩壞都會壓低 DO₂。這也解釋了一個常被忽略的事實:單純輸液或單純升壓並不一定改善組織供氧——如果病人同時嚴重貧血,把血壓拉高卻沒有補足攜氧的血紅素,組織照樣缺氧。
正常情況下,DO₂ 遠大於 VO₂,組織只萃取約 25% 的供氧,留有充裕的安全餘裕。當 DO₂ 開始下降,身體先靠「提高萃取率」維持 VO₂ 不變(這段稱為供應獨立 supply-independent)。但 DO₂ 一旦跌破某個臨界點(critical DO₂),萃取率再也補不上,VO₂ 就開始隨 DO₂ 一起下滑——細胞被迫轉入無氧代謝。這個轉折點,正是休克從「代償」走向「失代償」的生理分界。
乳酸與鹼缺失:細胞在求救的化學訊號
既然血壓會騙人,臨床上更倚重直接反映細胞代謝狀態的生化指標,其中最重要的就是乳酸(lactate)。
當組織缺氧、被迫進行無氧糖解,丙酮酸(pyruvate)無法進入粒線體完成有氧代謝,轉而還原成乳酸堆積。因此血中乳酸升高,是組織層級缺氧的客觀指紋。臨床研究反覆顯示,初始乳酸值與重症病人的死亡率高度相關,而乳酸清除率(lactate clearance)——亦即治療數小時後乳酸下降的幅度——更是反映復甦是否有效的關鍵動態指標。乳酸持續不降,常意味著組織灌流仍未恢復。
另一個互補指標是鹼缺失(base deficit),來自血液氣體分析,反映代謝性酸中毒的嚴重程度。在創傷病人身上,鹼缺失惡化往往早於血壓變化,是隱性出血的敏感警訊。
要提醒的是,乳酸升高不全然等於缺氧。劇烈運動、痙攣、某些藥物(如腎上腺素本身)與肝功能不良都可能影響乳酸。臨床判讀必須結合整體脈絡——但在急症情境下,看到一個外表還算穩定卻乳酸偏高的病人,經驗豐富的醫師會立刻提高警覺,因為那是身體在血壓崩潰前先送出的求救訊號。
四型休克:用機制分類,才能對症下藥
把休克依血流動力學機制分為四大類,是急重症診斷的骨幹。分類的意義不在背誦,而在於每一類的治療方向截然不同,甚至彼此衝突。
- 低血容性休克(Hypovolemic):血液或體液大量流失(出血、嚴重脫水、燒傷)。前負荷(preload)不足,核心處置是補充容量與止血。
- 心因性休克(Cardiogenic):幫浦本身故障(大面積心肌梗塞、嚴重心律不整、心衰竭末期)。心臟打不動,此時若大量灌注液體反而會淹沒已經衰竭的心臟,造成肺水腫——這正是與低血容性休克治療方向相反之處。
- 阻塞性休克(Obstructive):循環的「管路」被機械性堵住(張力性氣胸壓迫、心包填塞、大塊肺栓塞)。輸液升壓都只是治標,必須解除阻塞——例如針對張力性氣胸做胸腔減壓——病情才會逆轉。
- 分布性休克(Distributive):血管張力崩解、血液「分布」異常,總血量未必減少但血管過度擴張(敗血症、過敏性休克、神經性休克)。核心問題是血管阻力過低,除了輸液,常需血管收縮劑(vasopressor)把張力拉回來。
臨床上四型可能並存(例如嚴重敗血症合併心肌抑制),這正是急重症的難處:先用快速床邊評估與超音波辨明主要機制,才能選對處置。把心因性休克當低血容性休克猛灌水,或把張力性氣胸當敗血症拼命升壓,都會把病人推向更糟的境地。
流體反應性:該不該繼續給水?
休克復甦最核心、也最容易被誤用的工具就是「輸液」。傳統做法是「血壓低就快速灌注大量晶體液」,但現代重症醫學對此已大幅修正,核心問題變成:這個病人到底還會不會對輸液有反應?
這裡的關鍵概念是法蘭克—史達林曲線(Frank–Starling curve):心臟的每跳輸出量會隨前負荷(心室充盈)增加而上升,但曲線會逐漸平緩。病人若位於曲線陡升段,多給液體能有效提高心輸出量(即「有流體反應性」);若已在平台段,再灌液體不但無益,反而造成組織水腫、肺水腫、稀釋性凝血病變等傷害。
因此床邊評估「流體反應性(fluid responsiveness)」成為現代復甦的重要技能,方法包括被動抬腿試驗(passive leg raise,把下肢血液暫時回送、觀察心輸出量是否上升)、輸液衝擊後再評估、以及超音波觀察下腔靜脈隨呼吸的塌陷度等動態指標。其精神是用最小必要的液體達到灌流目標,而非盲目灌到血壓正常為止——過度復甦(over-resuscitation)本身就是一個獨立的傷害來源。
損害控制復甦:對創傷出血的範式轉移
過去數十年,創傷出血的處置思維發生了根本性轉變,集中體現在「損害控制復甦(damage control resuscitation)」這套概念上,其中兩個觀念尤其值得理解。
第一是容許性低血壓(permissive hypotension)。對於尚未止血的活動性出血病人,過去會積極把血壓灌到正常;但現代觀念認為,在止血完成前,刻意把血壓維持在「剛好能灌注重要器官」的較低水準(例如收縮壓約 80–90 mmHg,特定族群除外),反而能避免把好不容易形成的血栓「沖開」、減少稀釋凝血因子。這背後是對「凝血」與「灌流」之間張力的精細權衡。
第二是創傷誘發凝血病變(trauma-induced coagulopathy)與「致命三角(lethal triad)」。嚴重創傷會同時引發低體溫(hypothermia)、酸中毒(acidosis)與凝血功能障礙(coagulopathy),三者互相惡化形成死亡螺旋——酸中毒抑制凝血酶活性,低體溫讓血小板功能下降,而出血又加重前兩者。因此現代創傷復甦強調早期輸注血液製品(紅血球、血漿、血小板按接近生理比例給予)、積極保溫、並使用抗纖維蛋白溶解藥物(如 tranexamic acid)——目標是同時對抗三角的三個頂點,而不只是「補水撐血壓」。
看一個例子
讓我們回到開頭那位車禍的年輕女性,看看進階思維如何改變處置。
初評:意識清楚、血壓 118/76、心跳 122、皮膚濕冷、乳酸 5.8。若只看血壓,她「正常」;但心搏過速、末梢灌流不佳加上高乳酸,三者拼起來是典型的代償期低血容性休克——身體正在用交感活化把血壓硬撐住。
醫師沒有被血壓誤導,立刻啟動評估:超音波發現腹腔有游離液體,提示腹內出血(這是低血容性,不是分布性,方向是止血而非升壓)。團隊不是「灌爆晶體液把血壓沖更高」,而是採容許性低血壓策略——維持足以灌注腦與腎的最低血壓,同時啟動大量輸血方案(血液製品按比例給)、給予 tranexamic acid、並積極保暖以對抗致命三角。最關鍵的決定是:盡快送進手術室控制出血源,因為對活動性出血,任何復甦都只是替真正的止血爭取時間。
對照之下,若這是一位心因性休克病人(例如大面積心肌梗塞導致幫浦衰竭),同樣的「快速大量輸液」會直接造成肺水腫、讓病人更喘更缺氧。同樣是低血壓與休克,機制不同,處置幾乎相反——這就是為什麼進階急症的核心是「先辨機制,再談處置」。
動手試試
不需要任何儀器,你也可以練習「不被血壓綁架」的休克思維。試著針對下面三個情境,先判斷最可能的休克類型,再說出處置方向:
- 老人腹瀉三天、皮膚乾癟、血壓偏低、心跳快——最可能是低血容性(脫水),方向是補液。
- 蜂螫後數分鐘全身起疹、喉嚨緊、血壓驟降——分布性(過敏性休克),方向是肌肉注射腎上腺素加輸液。
- 胸痛病人突然血壓崩、頸靜脈怒張、一側呼吸音消失——阻塞性(疑張力性氣胸),方向是緊急胸腔減壓,而非一味升壓。
你會發現,真正的鑑別不在「血壓多少」,而在「血壓低的背後,是哪一條機制壞掉了」。能把這層因果講清楚,就已經跨過了入門到進階的門檻。
重點回顧
- 休克的本質是細胞缺氧,不是血壓低:強大的代償機制能在失血達 30% 時仍維持正常血壓,等血壓崩落往往已是失代償晚期。
- 氧氣供應由 DO₂ = 心輸出量 × 攜氧含量決定:心輸出量、血紅素、血氧飽和度任一崩壞都會缺氧,單純升壓或單純輸液未必改善組織供氧。
- 乳酸與鹼缺失是細胞層級的客觀警訊:乳酸清除率反映復甦成效,常在血壓變化前就揭露隱性休克。
- 四型休克機制不同、處置甚至相反:心因性與低血容性對「輸液」的反應背道而馳,阻塞性必須先解除機械阻塞。
- 現代復甦講究流體反應性與損害控制:用最小必要液體達標、容許性低血壓避免沖開血栓、並對抗低體溫—酸中毒—凝血病變的致命三角。
深入探討(研究所視角)
對有志於急重症醫學或臨床研究的讀者,以下幾個面向值得進一步探索。
微循環與大循環的脫鉤(hemodynamic coherence)。 我們在床邊監測的多半是大循環指標(血壓、心輸出量),但細胞真正仰賴的是微循環(microcirculation)的灌流。敗血症研究發現一個耐人尋味的現象:即使把大循環指標「治療到正常」,微血管層級的灌流障礙仍可能持續存在,組織照樣缺氧——這稱為大小循環的「不一致(loss of hemodynamic coherence)」。手持式顯微影像技術(如 sidestream dark field imaging)讓研究者得以直接觀察舌下微循環,正在改變我們對「復甦終點」的理解:把血壓拉正常,不等於把組織救活。
復甦終點之爭與目標導向治療。 早期目標導向治療(Early Goal-Directed Therapy, EGDT)曾以中心靜脈血氧飽和度等指標設定明確復甦目標,影響深遠;但後續多中心隨機試驗(ProCESS、ARISE、ProMISe)發現,嚴格遵循固定流程未必優於有經驗醫師的常規照護。這場辯論的價值在於提醒我們:復甦不是照表操課,而是依個別病人動態調整——這也呼應了入門篇 TTM 爭議所揭示的「臨床實證持續演化」本質。
血管加壓素與升壓策略的精細化。 分布性休克的升壓劑選擇(如正腎上腺素為一線、血管加壓素 vasopressin 作為輔助、必要時加入其他藥物)背後是受體生理與劑量反應的精細權衡。何時該補液、何時該升壓、兩者比例如何,是 ICU 每天面對的最佳化問題,也是機器學習輔助決策(如以強化學習推薦升壓/輸液策略)近年積極切入的研究場域——但其安全性與可解釋性仍在嚴格檢驗中。
從生理到資料科學。 連續監測產生的高頻波形(動脈壓波形、心率變異)蘊含豐富的代償狀態資訊。如何從這些訊號中提早數十分鐘預測失代償(例如預測即將發生的低血壓事件),是急重症與資料科學交會的前沿。這也是 Educational Omics 框架中 PhysioNeuromics 維度在臨床端的延伸:生理訊號不只反映學習狀態,更能在加護病房裡提早洩漏一個身體即將崩潰的時刻。
最後再次提醒,本文旨在建立機制層次的理解,不能取代專業訓練與臨床判斷。真正的休克辨識與復甦能力,需要在合格的臨床訓練與實作中,把這些原則內化為臨場的判斷反射。