
一杯葡萄柚汁,為什麼能讓一顆「安全劑量」的藥變成中毒劑量?
超越半衰期,用清除率、分布容積與非線性動力學看懂劑量設計、藥物交互作用與受體佔據的量化邏輯。
一杯葡萄柚汁,為什麼能讓一顆「安全劑量」的藥變成中毒劑量?
在入門篇裡,我們知道藥物進入身體後要走過吸收、分布、代謝、排泄(ADME),也知道血中濃度的高低決定療效與毒性。但臨床上真正令人頭痛的問題往往不是「這顆藥有沒有效」,而是更精確的版本:到底要給多少?多久給一次?第一劑要不要加重?腎臟壞掉的病人劑量要打幾折?兩種藥一起吃,血中濃度會不會悄悄爆表?
這些問題沒辦法靠直覺回答,它們是可以計算的。臨床藥理學最迷人也最被低估的一面,就是它其實是一門用幾個參數、幾條方程式,把「人體這個黑盒子」量化到能預測藥物濃度的工程學。本篇假設你已經讀過入門篇、知道 PK/PD 與 ADME 的大方向,我們要往下鑽:把抽象的「半衰期」拆解成它背後真正的兩個物理量,看懂劑量設計的數學,並理解為什麼有些藥物會違反「線性」直覺、突然從安全變致命。
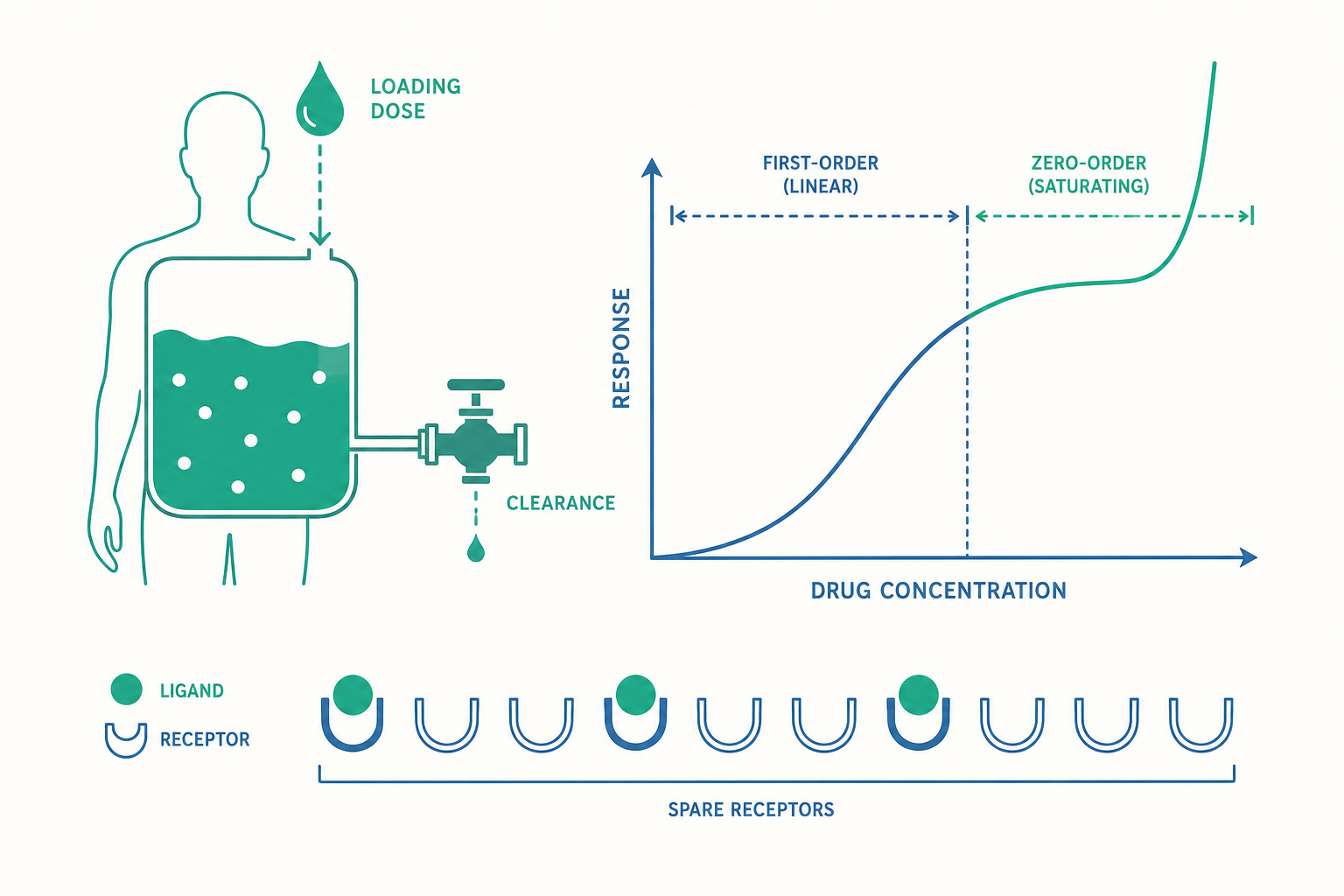
半衰期其實不是基本參數:清除率與分布容積才是
入門篇用「半衰期(half-life, t½)」描述藥物消除的速度,這對日常理解很夠用。但在進階視角下,半衰期是一個衍生(derived)參數——它本身不能獨立決定病人發生什麼事,而是由兩個更根本的量算出來的。
第一個是清除率(clearance, CL):單位時間內,身體能「徹底清乾淨」的血漿體積(單位通常是 L/h 或 mL/min)。注意它衡量的不是「清掉多少藥」,而是「清乾淨了多少體積的血漿」。清除率主要反映肝臟代謝與腎臟排泄的總能力,是決定維持劑量(maintenance dose)的關鍵。
第二個是分布容積(volume of distribution, Vd):一個「表觀(apparent)」的體積,定義為「體內藥物總量÷血漿中藥物濃度」。它不是真的某個器官的容積,而是一個比例常數,告訴你藥物有多「願意」離開血液、躲進組織。一個高度親脂、會大量蓄積在脂肪與組織的藥,可能算出比全身體液還大好幾倍的 Vd(聽起來很荒謬,但這正是「表觀」二字的意思——它是個換算因子,不是解剖學體積)。Vd 是決定負荷劑量(loading dose)的關鍵。
三者的關係可以寫成一條核心方程式:
$$t_{½} = \frac{0.693 \times V_d}{CL}$$
這條式子立刻顛覆了一個常見迷思。半衰期變長,不一定代表「身體清除藥物的能力變差」。 想像兩個情境:
- 病人 A 因為腎衰竭,CL 下降一半 → t½ 變兩倍。這是「清除變差」。
- 病人 B 因為脫水或體液變化導致 Vd 增加一倍,CL 完全正常 → t½ 也變兩倍。但他的清除能力一點都沒變。
兩人都「半衰期變兩倍」,臨床意義卻完全不同。如果只盯著半衰期,你會把 B 誤判成清除功能受損。真正驅動劑量決策的是 CL 和 Vd,半衰期只是它們的影子。 這是初學者與進階者的分水嶺之一。
劑量怎麼算:負荷劑量管「多快到位」,維持劑量管「穩在哪裡」
理解了 CL 與 Vd,劑量設計就從「憑經驗」變成「可推導」。臨床上要做兩件事:先快速把濃度拉到有效範圍(負荷劑量),再用穩定的速率維持它(維持劑量)。
負荷劑量(loading dose):用 Vd 填滿這個「容器」
如果你想讓血漿濃度一步到位達到某個目標值(target concentration, $C_{target}$),需要的劑量就是「想要的濃度」乘上「藥物會散布的體積」:
$$\text{負荷劑量} = \frac{V_d \times C_{target}}{F}$$
(F 是生體可用率,靜脈注射時 F=1。)這也解釋了入門篇提過的現象:很多藥若只靠每天的維持劑量慢慢累積,要等 4~5 個半衰期才達穩態——對半衰期長的藥可能是好幾天。如果病人正在敗血症、癲癇重積或嚴重心律不整,等不了這麼久,就先給一劑負荷劑量「一次填滿容器」,再接維持劑量。
維持劑量(maintenance dose):用 CL 補上漏掉的量
到了穩態(steady state),一個漂亮的物理直覺成立:進去的速率 = 出來的速率。藥物排出的速率等於「清除率×濃度」,所以要維持某個穩態濃度 $C_{ss}$,給藥速率必須剛好補上這個損失:
$$\text{給藥速率} = CL \times C_{ss}$$
請注意這條式子裡沒有 Vd,也沒有半衰期。穩態濃度只由「給藥速率」和「清除率」決定。這帶出一個關鍵臨床推論:腎功能不佳的病人之所以要減量,是因為他們的 CL 下降,若維持速率不變,$C_{ss}$ 會等比例升高、可能進入中毒區。 而 Vd 改變只影響「多快達到穩態」與「波動幅度」,不影響穩態的平均高度。把劑量決策拆成「負荷靠 Vd、維持靠 CL」這兩條清楚的軌道,是臨床藥理的核心邏輯。
當直覺失效:非線性(飽和)藥物動力學
到目前為止我們默默假設了一件事:藥物濃度與劑量成正比,半衰期是固定的——這叫一階(first-order)動力學,是大多數藥物在治療濃度下的行為。在一階動力學裡,「每單位時間清除掉固定比例」,所以你把劑量加倍,穩態濃度也大致加倍,很線性、很乖。
但有一小群極為重要的藥物會背叛這個直覺。原因是它們的代謝依賴會被飽和的酵素系統。當血中濃度低時,酵素還有餘裕,清除像一階動力學一樣按比例進行;可是一旦濃度高到把代謝酵素「灌滿」,清除速率就達到上限、不再隨濃度上升——這時變成零階(zero-order)動力學:每單位時間只能清掉固定的量,而不是固定的比例。
這個轉變由 Michaelis-Menten 方程式描述,整體稱為非線性(飽和)藥物動力學。它在臨床上的後果非常凶險:在飽和點附近,劑量只增加一點點,穩態濃度可能暴增到數倍,因為身體的清除能力已經頂到天花板,多給的藥幾乎全部「滯留」。
三個經典例子值得記住:
- 苯妥英(phenytoin,抗癲癇藥):教科書級的非線性藥物。劑量從每日 300 mg 微調到 400 mg,血中濃度可能不是上升 33%,而是翻倍進入毒性區,導致眼球震顫、運動失調。
- 乙醇(ethanol):你的身體大約以固定速率代謝酒精(典型約每小時一個「標準杯」的量),與血中酒精濃度幾乎無關——這正是零階動力學。所以喝越快、超過肝臟固定處理速率,血中濃度就直線飆高。
- 阿斯匹靈(aspirin)高劑量:低劑量呈一階,高劑量時代謝路徑飽和、轉為零階,是水楊酸中毒難以預測的原因之一。
對這類藥物,「半衰期」這個詞幾乎失去意義——因為它根本不是固定的,會隨濃度變化。臨床上必須用治療藥物監測(therapeutic drug monitoring, TDM)抽血量濃度,而不能單靠劑量推算。
看一個例子:兩種藥物,同樣加量 50%,命運天差地遠
設想一位醫師要把病人的劑量從 100 單位調到 150 單位(增加 50%)。
- 藥物甲走一階動力學(如多數抗生素、多數降血壓藥):穩態濃度大致也上升約 50%,平順可預期,落在安全範圍內。
- 藥物乙走非線性動力學、且目前濃度已接近酵素飽和點(如苯妥英):同樣加 50% 的劑量,穩態濃度可能上升 150%、200% 甚至更多,直接衝進毒性區。
同樣的「加量 50%」這個動作,在甲身上是微調,在乙身上可能是醫療事故。這就是為什麼理解一個藥物屬於線性還是非線性,往往比記住它的半衰期更救命。本文為醫學知識說明,並非個人醫療建議,任何用藥調整都應由醫師或藥師依個別情況評估。
藥物交互作用的機制:回到開頭那杯葡萄柚汁
現在我們有足夠的工具,可以精確回答開頭的問題了。入門篇提過葡萄柚汁會抑制肝臟的 CYP3A4 酵素,這裡我們看它在數學上意味著什麼。
許多藥物(某些降血脂的 statin、某些鈣離子通道阻斷劑、某些免疫抑制劑)主要靠 CYP3A4 代謝。當葡萄柚汁中的呋喃香豆素(furanocoumarins)不可逆地抑制腸道與肝臟的 CYP3A4:
- 該藥的清除率 CL 下降。
- 由前面的維持劑量公式 $C_{ss} = \text{給藥速率} / CL$,CL 變小 → 穩態濃度成比例上升。
- 對某些藥物,腸壁 CYP3A4 被抑制還會減少首渡代謝、使生體可用率 F 上升,雙重夾擊讓血中濃度進一步飆高。
於是,一個原本完全在安全範圍內的「標準劑量」,因為清除能力被外力砍半,穩態濃度可能翻倍,把藥物推進治療指數(therapeutic index)的危險區——對某些 statin 而言,這意味著橫紋肌溶解的風險上升。這不是玄學,而是 CL、F 這幾個參數被改動後,公式必然導出的結果。
交互作用大致可分兩類機制,現在你能看穿它們的本質:
- 藥物動力學交互作用(PK interaction):一個藥改變另一個藥的 ADME,最終改變的是血中濃度。CYP 抑制劑(如葡萄柚汁、某些抗黴菌藥)讓濃度升高;CYP 誘導劑(如某些抗癲癇藥、聖約翰草)加速代謝、讓濃度降低、療效打折。
- 藥效學交互作用(PD interaction):兩個藥血中濃度都沒變,但在作用部位產生加成、協同或拮抗。例如同時服用兩種會抑制中樞神經的藥物(鎮靜劑加酒精),呼吸抑制效果相加,這是濃度以外的危險。
把「交互作用」拆成「它到底動了哪個參數」——是 CL?是 F?還是受體層次的效果疊加?——你就從死背交互作用清單,升級成能推理新組合風險的思考者。
受體佔據與「備用受體」:為什麼有些藥不必佔滿就全力運轉
入門篇用「鑰匙與鎖」說明致效劑與拮抗劑,並提到親和力與內在活性。進階一步,我們要量化「到底要佔住多少把鎖,才能產生多少效果」——這就是受體佔據理論(receptor occupancy theory)。
最素樸的假設是「效果與被佔據的受體比例成正比」:佔了一半受體就有一半效果。但實驗常常打臉這個假設。許多系統存在備用受體(spare receptors / receptor reserve)現象:致效劑只要佔據一小部分受體(有時不到 10%),就能引發最大反應(Emax)。原因是受體下游的訊號放大級聯非常有效率,少數被活化的受體就足以把訊號推到飽和。
這個概念有兩個重要的臨床推論:
- 一個藥物的效價強度(potency, EC₅₀)可能與它的受體親和力(Kd)不一致——因為有備用受體時,達到半數效果所需佔據的受體遠少於半數,使藥物「看起來」比親和力預期的更強。
- 它解釋了某些不可逆拮抗劑的行為:當你用不可逆拮抗劑逐步「報廢」受體,一開始因為有備用受體,致效劑還能達到最大效果(曲線只是右移);但當報廢的受體超過備用量,最大效果才開始被壓低。
備用受體提醒我們:「佔據率」與「效果」之間隔著一整套訊號放大機制,不能直接畫等號。這也是為什麼藥效學不能只看結合數據,必須測量真正的功能反應。
重點回顧
- 半衰期是衍生參數,真正的基本量是清除率(CL)與分布容積(Vd),關係為 $t_{½} = 0.693 \times V_d / CL$。半衰期變長可能源於 CL 下降「或」Vd 上升,兩者臨床意義完全不同。
- 劑量設計分兩軌:負荷劑量由 Vd 決定(填滿容器、快速到位),維持劑量/穩態濃度由 CL 決定($C_{ss} = $ 給藥速率 $/ CL$)。腎功能差要減量,是因為 CL 下降。
- 非線性(飽和)動力學是直覺的陷阱:當代謝酵素被灌滿,清除從一階轉為零階,劑量小增可能讓濃度暴增。苯妥英、乙醇、高劑量阿斯匹靈是經典例子,需靠治療藥物監測(TDM)把關。
- 交互作用的本質是參數被改動:PK 交互作用改變的是血中濃度(CYP 抑制讓濃度升、誘導讓濃度降),PD 交互作用則是作用部位的效果疊加。葡萄柚汁透過抑制 CYP3A4 同時降低 CL、提高 F,使濃度雙重上升。
- 受體佔據不等於效果:備用受體現象使少數受體被佔據即可達 Emax,導致效價強度與親和力可能分離,並影響不可逆拮抗劑的劑量-反應曲線形狀。
深入探討(研究所視角)
把臨床藥理的量化思維再往研究前沿推一層,會看到三個正在重塑這個領域的方向。
從個體參數到族群模型:非線性混合效應建模。 前文的 CL 與 Vd,在真實病人身上不是單一數字,而是有族群平均值、也有個體間變異(inter-individual variability)與個體內變異(residual variability)。族群藥物動力學(population PK)運用非線性混合效應模型(nonlinear mixed-effects modeling, NLME),從臨床上稀疏、不規則的抽血資料中同時估計這些層次的變異,並找出哪些共變項(covariates)——體重、年齡、腎絲球過濾率、基因型——能解釋變異。這讓「根據病人特徵推算個人化劑量」從經驗法則變成統計推論,是現代藥物仿單上劑量建議的科學基礎。下一步是貝氏估計(Bayesian forecasting):結合族群先驗與病人自身的一兩筆血中濃度,即時回推該病人專屬的 CL/Vd,動態調整劑量,正在重症與抗生素治療領域逐步落地。
機制取向的整合:PBPK 與定量系統藥理學。 傳統 PK 把身體當成一兩個「房室(compartment)」的抽象黑盒子。生理基礎藥物動力學(physiologically-based pharmacokinetics, PBPK)則反其道而行,用真實的器官血流量、組織體積與分配係數,把身體建成由多個解剖學房室串接的系統,能在「沒有臨床資料」的情況下預測藥物在特定器官的濃度、外推到兒童或孕婦等難以試驗的族群。再往上一層的定量系統藥理學(QSP),把分子訊號網絡、細胞反應與整體生理整合進同一個計算模型,試圖回答「為什麼這個標的的藥在某些病人無效」這類機制問題。這種多尺度、多模態的整合思維,與 Uedu 倡議的 Educational Omics 框架在方法論上同源——都相信唯有把分子、生理、行為等多層次資料納入同一架構,才能逼近真實系統的複雜性。
精準藥理與下一代標的。 入門篇談過藥物基因體學(pharmacogenomics)如何解釋代謝差異;進階視角會看到它正與兩股力量匯流。其一是生物製劑與標的遞送:單株抗體、抗體-藥物複合體(ADC)與細胞療法,把「小分子鑰匙」擴展為能精準導引、具備自己一套 PK 特性(如標的介導的藥物處置, target-mediated drug disposition)的大分子,傳統線性 PK 模型必須重寫。其二是人工智慧驅動的藥物設計:機器學習正被用於預測 ADMET 性質、辨識新標的、優化分子結構,壓縮藥物開發的時間與成本。然而,無論工具多先進,所有努力最終都要回到帕拉塞爾蘇斯五百年前那條界線——療效與毒性之間、那條因人而異的劑量分界。對有志深入的學生而言,把 CL、Vd、線性與非線性、受體佔據這些原理真正內化成可推理的工具,正是站上這些前沿、而不被新名詞淹沒的根基。