
為什麼同樣一顆止痛藥,有人吃半顆就有效,有人吃兩顆還喊痛?
從藥物動力學(PK)與藥效學(PD)出發,理解一顆藥從吸收、分布、代謝、排泄到與受體結合作用的完整旅程,以及劑量、個體差異與精準用藥。
為什麼同樣一顆止痛藥,有人吃半顆就有效,有人吃兩顆還喊痛?
想像三位學生同時頭痛,各自吞下一顆 500 毫克的乙醯胺酚(acetaminophen,普拿疼的主成分)。一小時後,第一位覺得頭痛明顯緩解,第二位只覺得「好像有比較好」,第三位則幾乎沒感覺。藥丸成分一模一樣、劑量一模一樣,為什麼結果差這麼多?
答案藏在「藥物進入身體之後到底發生了什麼事」這個問題裡。一顆藥從你吞下去的那一刻起,要先溶解、被腸道吸收、進入血液、分布到全身、找到它該作用的目標、產生效果,最後被代謝、排出體外。這一連串歷程的每一步,都會因為個人的體重、肝臟酵素、基因、共服藥物甚至飲食而改變。藥理學(pharmacology),就是研究「藥物與生命系統如何互相影響」的學問。它不只關心「這顆藥能治什麼病」,更追問「為什麼能治、怎麼作用、在誰身上會怎樣」。
藥理學的兩個核心問題:身體對藥、藥對身體
藥理學最經典的框架,是把藥物與身體的互動拆成兩個方向,像照鏡子一樣對稱:
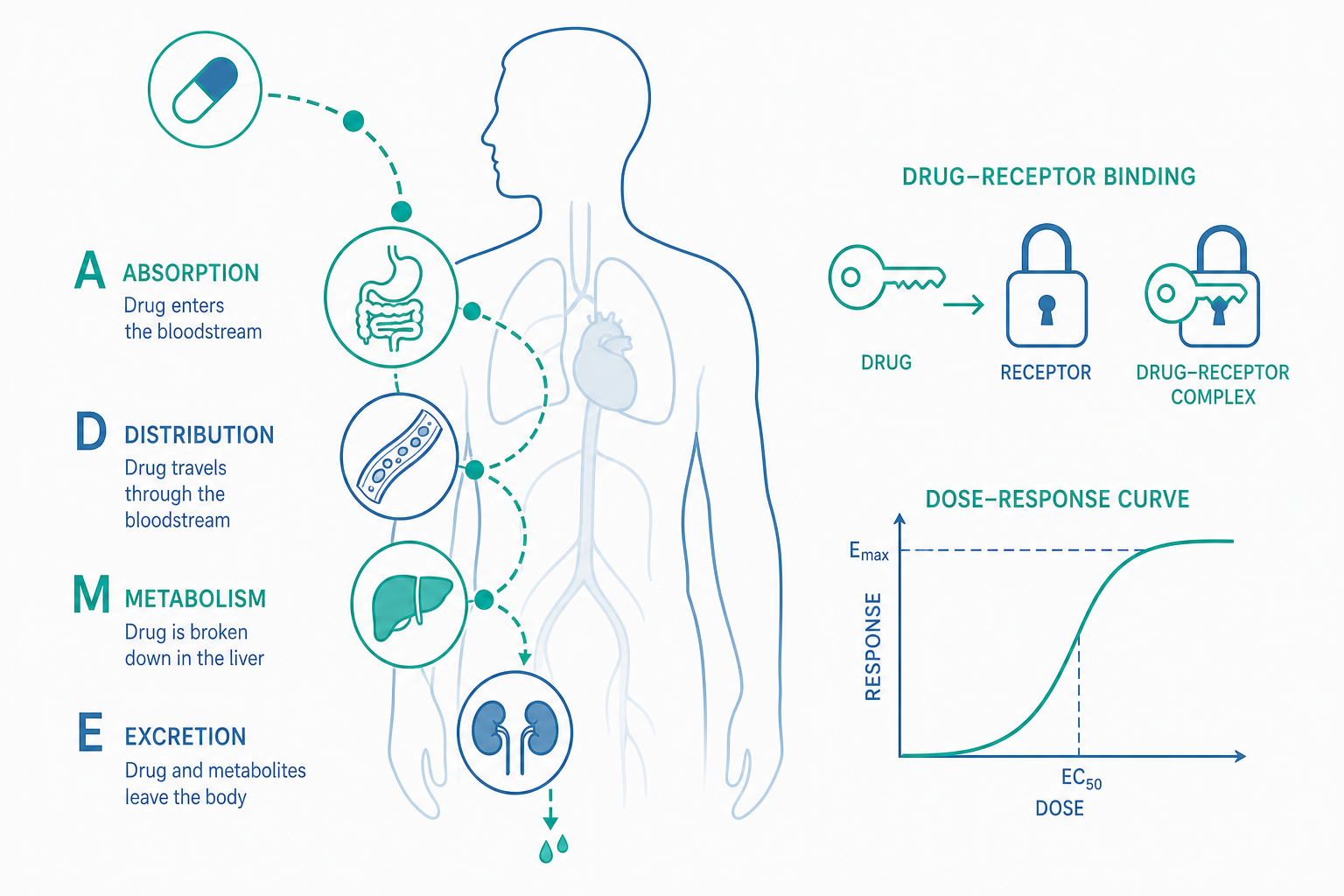
- 藥物動力學(pharmacokinetics, PK):身體對藥物做了什麼。也就是藥物的吸收(absorption)、分布(distribution)、代謝(metabolism)、排泄(excretion),常以英文字首縮寫為 ADME。這決定了血液中的藥物濃度隨時間如何變化。
- 藥效學(pharmacodynamics, PD):藥物對身體做了什麼。也就是藥物在作用部位產生效果的機制,包括它如何與受體(receptor)結合、引發何種反應、劑量與效果之間的關係。
用一句話記憶:PK 是「藥物在哪裡、有多少」,PD 是「藥物做了什麼、做得多強」。 開頭那三位頭痛的學生,差異很可能就出在 PK——他們的肝臟代謝乙醯胺酚的速度、體重造成的分布容積不同,使得相同劑量在血中達到的有效濃度不一樣。
理解這兩個方向,是理解一切藥物的鑰匙。接下來我們先走一趟藥物在體內的旅程(PK),再看藥物如何「按下開關」(PD)。
藥物的體內旅程:ADME 四部曲
吸收(Absorption):藥怎麼進入血液
口服是最常見的給藥途徑,但也是最「曲折」的一條路。藥錠在胃腸道溶解後,必須穿過腸壁細胞進入血液。這裡有一個關鍵概念叫首渡效應(first-pass effect):從腸道吸收的血液會先經由肝門靜脈進入肝臟,肝臟的酵素可能在藥物到達全身循環之前,就先代謝掉一大部分。這就是為什麼某些藥物口服劑量必須遠高於靜脈注射劑量——因為一部分還沒發揮作用就被肝臟「攔截」了。
衡量這一步效率的指標是生體可用率(bioavailability, F):實際進入全身循環的藥量佔給藥總量的比例。靜脈注射的 F 定義為 100%,因為藥物直接進血管,不經過吸收與首渡的損耗;口服藥的 F 則因藥物特性而異,可能高達九成,也可能低到只剩一兩成。
分布(Distribution):藥跑到哪裡去
進入血液後,藥物會隨循環分布到各組織。這裡有兩個重要因素。其一是血漿蛋白結合:許多藥物會與血中的白蛋白(albumin)結合,只有「游離型(free)」的藥物才能離開血管、產生作用。其二是血腦障壁(blood-brain barrier, BBB):腦部微血管有緊密的內皮屏障,脂溶性高的藥物較容易穿越進入中樞神經系統,水溶性高、分子大的藥物則進不去——這解釋了為什麼有些止痛藥會讓人想睡(作用於腦),有些則不會。
代謝(Metabolism):身體如何「拆解」藥物
代謝主要發生在肝臟,目的是把脂溶性藥物轉變成較水溶性、易於排出的形式。其中最重要的角色是細胞色素 P450 酵素系統(cytochrome P450, CYP),尤其是 CYP3A4 與 CYP2D6 等亞型,負責代謝市面上大量藥物。
CYP 系統是藥物交互作用的重災區。舉例來說,葡萄柚汁會抑制 CYP3A4,使某些降血壓藥或降血脂的 statin 類藥物代謝變慢、血中濃度升高,可能導致副作用增強——這不是民間傳說,而是有明確機制的藥物-食物交互作用。反過來,有些藥物會誘導(induce)CYP 活性,加速其他藥物的代謝,反而降低療效。
排泄(Excretion):藥物如何離開
最終藥物(或其代謝產物)主要經由腎臟隨尿液排出,部分經膽汁、糞便、汗液或呼氣排出。腎功能不佳的患者,藥物排泄變慢、容易在體內累積,因此許多藥物需依腎功能調整劑量。
描述藥物消除速度的核心指標是半衰期(half-life, t½):血中藥物濃度下降到一半所需的時間。半衰期決定了給藥頻率——半衰期短的藥需要一天多次服用,半衰期長的藥一天一次即可。一般而言,經過約 4 到 5 個半衰期後,藥物在體內大致清除完畢;同樣地,規律服藥約 4 到 5 個半衰期後,血中濃度會達到穩定的穩態(steady state)。
藥物如何作用:受體、致效劑與拮抗劑
走完 PK 的旅程,藥物終於抵達作用部位,這時換 PD 登場。大多數藥物不是「憑空」產生效果,而是與身體的特定標的分子(target)結合——最常見的是受體(receptor),此外還有酵素、離子通道、轉運蛋白等。
受體就像細胞表面或內部的「鎖」,藥物則像「鑰匙」。能與受體結合並啟動其生理反應的藥物稱為致效劑(agonist,又譯促效劑);能與受體結合卻不啟動反應、反而擋住天然訊號的稱為拮抗劑(antagonist)。
這個「鑰匙與鎖」的比喻可以帶出兩個更精細的概念:
- 親和力(affinity):藥物與受體結合的緊密程度——鑰匙插不插得進鎖孔。
- 內在活性/效能(efficacy):結合後能引發多強的反應——鑰匙能不能把鎖轉開。
致效劑兩者兼具:既能結合,又能轉開鎖。拮抗劑則有親和力(能塞進鎖孔)但沒有內在活性(轉不開鎖),於是它佔住位置,讓真正的鑰匙進不來。介於兩者之間的還有部分致效劑(partial agonist),能結合並引發部分反應,但即使佔滿所有受體也達不到完全致效劑的最大效果。
看一個例子:β 受體與兩類心臟用藥
心臟與血管上分布著腎上腺素能受體(adrenergic receptor),其中 β₁ 受體主要在心臟。當身體分泌腎上腺素、與 β₁ 受體結合時,會使心跳加快、收縮力增強——這是「戰或逃」反應的一部分。
現在來看兩類藥:
- β 致效劑(如某些急救或氣喘用藥)模擬腎上腺素的作用,能在需要時加快心跳、擴張支氣管。
- β 阻斷劑(β-blocker,β 拮抗劑)則相反,它佔住 β₁ 受體、擋住腎上腺素的訊號,使心跳減慢、心臟負荷減輕。這類藥常用於高血壓、心律不整與某些心臟病的長期管理。
同一個受體,致效與拮抗就是兩個方向相反的治療策略。值得注意的是,受體有不同亞型(β₁ 主要在心臟、β₂ 主要在支氣管與血管),藥物對亞型的選擇性(selectivity)越高,越能精準作用、減少不必要的副作用。一個只擋 β₁ 的「心臟選擇性」阻斷劑,比起同時擋 β₁ 與 β₂ 的非選擇性藥物,較不易引發支氣管收縮——這對有氣喘病史的患者特別重要。
劑量與反應:為什麼「劑量決定毒性」
文藝復興時期的醫師帕拉塞爾蘇斯(Paracelsus)有一句名言常被引用:「萬物皆有毒,無物不毒;唯有劑量區分毒與藥。」 這句話精準點出藥理學的核心——同一種物質,劑量太低沒效果,劑量適中是良藥,劑量過高就是毒物。水喝太多會中毒,肉毒桿菌毒素微量卻能治病。
劑量-反應關係(dose-response relationship)描述的就是劑量增加時效果如何變化。典型的曲線呈 S 形:低劑量時效果隨劑量快速上升,到了一定程度後趨於平緩,因為受體幾乎都被佔滿,再加藥也無法增加效果(達到最大效應, Emax)。
兩個衍生概念很重要:
- 效價強度(potency):達到特定效果所需的劑量——所需劑量越低,效價越強。注意「效價強」不等於「效果好」,它只代表「用比較少的量就能達到同樣效果」。
- 治療指數(therapeutic index, TI):衡量藥物安全範圍的指標,通常以「產生毒性的劑量」與「產生療效的劑量」之比表示。TI 越大,安全範圍越寬;TI 越小(如某些抗凝血劑、強心劑),有效劑量與中毒劑量很接近,必須嚴密監測血中濃度。
動手試試:估算一次服藥後何時達穩態
假設某口服藥的半衰期是 6 小時,醫囑是每天服用。請試著推算:
- 一個半衰期(6 小時)後,單次劑量約剩多少?(答:剩約一半,50%)
- 大約幾小時後體內藥物可視為清除完畢?(答:約 4 至 5 個半衰期,即 24 至 30 小時)
- 若每天規律服用,大約何時血中濃度達到穩態?(答:同樣約 4 至 5 個半衰期,約 1 至 1.5 天)
這個簡單的推算解釋了一個常見的臨床現象:許多慢性病用藥(如部分降血壓藥、抗憂鬱藥)需要連續服用數天甚至數週,效果才會完全顯現——因為要等血中濃度累積到穩態。這也是為什麼不可因為「吃幾天沒感覺」就自行停藥或加量。本文僅為醫學知識說明,並非個人醫療建議,任何用藥調整都應諮詢醫師或藥師。
重點回顧
- 藥理學研究藥物與生命系統的雙向互動,可拆成「身體對藥」的藥物動力學(PK,即 ADME)與「藥對身體」的藥效學(PD)。
- ADME 四部曲——吸收、分布、代謝、排泄——決定血中藥物濃度。首渡效應與生體可用率影響口服藥的效率;肝臟 CYP 酵素是代謝與藥物交互作用的關鍵;半衰期決定給藥頻率與達到穩態的時間。
- 多數藥物透過與受體結合作用:致效劑啟動反應、拮抗劑擋住天然訊號、部分致效劑介於兩者之間。親和力決定「結不結合」,內在活性決定「引發多強反應」。
- 劑量決定一切:劑量-反應呈 S 形曲線,效價強度衡量所需劑量、治療指數衡量安全範圍。「唯有劑量區分毒與藥」。
- 個體差異真實存在:體重、肝腎功能、基因、共服藥物與飲食都會改變藥物的效果,這是精準用藥的基礎。
深入探討(研究所視角)
當我們把鏡頭拉近,藥理學會展開為一個橫跨分子生物學、系統生理學與資料科學的研究領域。
受體訊號傳遞的分子細節。 前文「鑰匙與鎖」的比喻在研究所層級需要更精緻的模型。以G 蛋白偶聯受體(G protein-coupled receptors, GPCR)為例——這是人體最大的受體家族,也是約三分之一上市藥物的標的。當前研究已超越「致效/拮抗」的二分法,進入偏向訊號(biased agonism / functional selectivity)的時代:同一個受體可偶聯到 G 蛋白與 β-arrestin 兩條下游路徑,而某些配體能「偏向」只活化其中一條。這帶來了治療上的想像——例如設計只走鎮痛路徑、避開呼吸抑制路徑的鴉片類藥物,可能改善其安全性。此外,受體的組成型活性(constitutive activity)概念也修正了傳統觀點:有些受體即使沒有配體也有基礎活性,於是出現了能降低基礎活性的反向致效劑(inverse agonist),這是經典模型無法解釋的。
藥物基因體學與精準醫療。 開頭三位學生的個體差異,在研究前沿被推進到藥物基因體學(pharmacogenomics)。CYP2D6 等代謝酵素具有顯著的基因多型性,個體可被分類為「慢代謝者」「中間代謝者」「快代謝者」與「超快代謝者」。一個經典案例是抗血小板藥 clopidogrel:它是前驅藥(prodrug),需經 CYP2C19 活化才有效,攜帶失能變異的患者活化不足、療效打折。如今臨床指引已開始建議在特定族群用藥前進行基因檢測,這正是「對的藥、對的劑量、給對的人」的實踐。
PK/PD 模型與系統藥理學。 現代藥物開發大量依賴數學建模與模擬。族群藥物動力學(population PK)運用非線性混合效應模型,從稀疏的臨床資料中估計參數的族群平均值與個體間變異,據以設計給藥方案。更進一步的定量系統藥理學(quantitative systems pharmacology, QSP),則嘗試把分子網絡、細胞反應與整體生理整合成計算模型,模擬藥物在複雜系統中的行為。這與 Uedu 所倡議的 Educational Omics 多模態資料整合精神相通:唯有把多層次資料納入同一框架,才能逼近真實的複雜性。
跨領域連結與未竟之問。 藥理學正與多個領域深度交織:與免疫學結合催生了單株抗體與免疫檢查點抑制劑等生物製劑,把「小分子鑰匙」擴展到「大分子精準導引」;與奈米科技結合發展出標靶藥物遞送系統,試圖讓藥物只在病灶釋放、減少全身副作用;與人工智慧結合則加速了藥物標的辨識與分子設計。然而核心的挑戰依舊:如何在療效與毒性之間找到那條由帕拉塞爾蘇斯點出、卻因人而異的界線?這個五百年前的提問,至今仍是藥理學研究的動力來源。對有志於此的學生而言,扎實理解 PK/PD 的基本原理,正是踏入這些前沿的必要起點。