
為什麼同一個 CSTR,開機時穩在低溫、運轉中卻會突然「跳」到高溫?
從放熱 CSTR 的多重穩態與點火–熄火遲滯,深入停留時間分布(RTD)數學與觸媒顆粒內 Thiele 模數的擴散–反應競賽,看真實反應器如何偏離理想方程。
為什麼同一個 CSTR,開機時穩在低溫、運轉中卻會突然「跳」到高溫?
你在入門篇學過:CSTR 的設計方程是一條乾淨的代數式,給定進料與速率定律,就能算出體積與轉化率。但如果你真的去操作一座放熱反應的 CSTR,會遇到一個課本代數式預測不到的怪現象——把冷卻水溫度緩緩調高,反應器溫度起初也緩緩上升,直到某一刻「啪」地一聲跳上去,轉化率從 20% 暴衝到 95%;而當你想退回來、把冷卻水溫度調低,它卻不照原路回去,要降到比剛才低得多的溫度才肯「熄火」掉回低轉化率。
這不是儀器故障,而是同一組進料條件下,反應器存在多重穩態(multiple steady states)。線性的代數直覺在此失靈,因為阿瑞尼斯方程把溫度塞進了指數,使「產熱」對溫度呈 S 形非線性,而「移熱」對溫度呈直線——兩條曲線可以相交三次。這篇進階篇要帶你越過入門的理想反應器手算,進入三個真實反應器的核心機制:多重穩態與點火–熄火(ignition–extinction)、真實流動的停留時間分布(RTD)數學,以及觸媒顆粒內擴散與反應的競爭(Thiele 模數)。每一個都是把入門那條漂亮方程「打碎、再依現實重建」的過程。
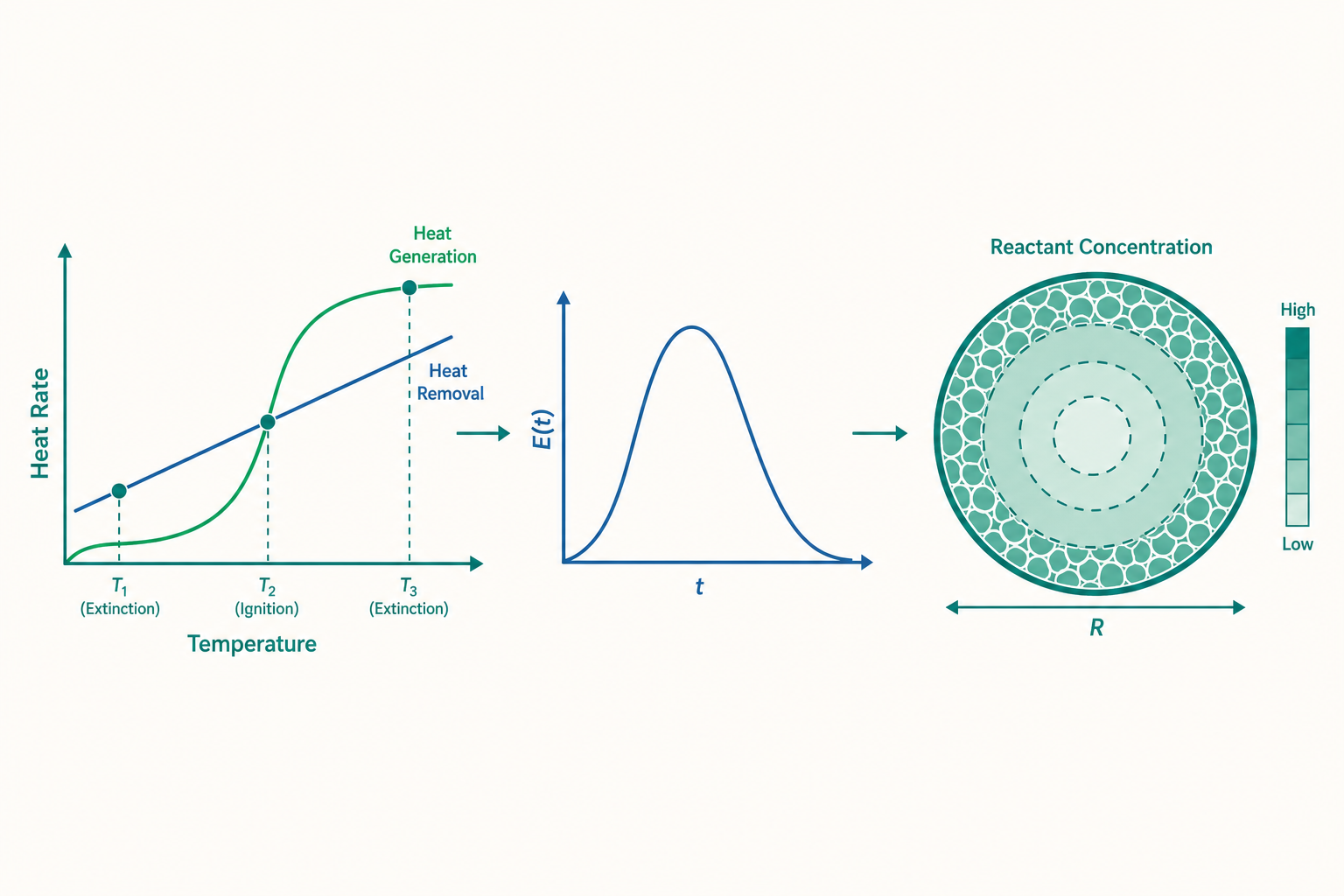
機制一:非絕熱 CSTR 的多重穩態
我們把入門的能量平衡推到底。考慮一個放熱一級反應 $A \to B$ 在 CSTR 中進行,穩態下要同時滿足莫耳平衡與能量平衡。莫耳平衡給出轉化率與溫度的關係(速率常數 $k(T)$ 走阿瑞尼斯):
$$X_A(T) = \frac{\tau\, k(T)}{1 + \tau\, k(T)}, \qquad k(T) = A\, e^{-E_a/(RT)}$$
把它代進能量平衡。我們定義兩個量:產熱速率(heat generated) $Q_g$ 與 移熱速率(heat removed) $Q_r$。
$$Q_g(T) = (-\Delta H_{\text{rxn}})\,\dot{n}_{A0}\,X_A(T) = (-\Delta H_{\text{rxn}})\,\dot{n}_{A0}\,\frac{\tau k(T)}{1+\tau k(T)}$$
$$Q_r(T) = \dot{n}_{\text{total}}\,C_p\,(T - T_{\text{in}}) + UA_{\text{c}}\,(T - T_{\text{c}})$$
關鍵在於兩條曲線的形狀。$Q_g(T)$ 因為 $k(T)$ 走指數、又被 $X_A$ 飽和到 1,整體是一條 S 形(sigmoidal) 曲線:低溫時幾乎貼地(反應太慢),中段陡升,高溫飽和(反應物已快耗盡)。而 $Q_r(T)$ 對 $T$ 是一條直線。穩態的條件是 $Q_g(T) = Q_r(T)$,也就是 S 形曲線與直線的交點。
一條直線最多能與 S 形曲線交於三點。當移熱直線的斜率與截距落在某個範圍內,就會出現三個穩態:
- 低溫穩態(熄火態):轉化率低,反應幾乎沒在跑。
- 中溫穩態(不穩定):數學上存在,但物理上不可達——任何微擾都會讓它滑向高溫或低溫態。
- 高溫穩態(點火態):轉化率高,反應旺盛。
動手試試:判斷穩態穩定性的斜率準則
哪個穩態穩定、哪個不穩,有一個漂亮的判據。在某個交點附近,如果溫度被擾動了一點點 $+\delta T$:
- 若此時產熱增加得比移熱還快($\dfrac{dQ_g}{dT} > \dfrac{dQ_r}{dT}$),多出來的熱會讓溫度繼續往上跑——這是不穩定的(正回饋)。
- 若移熱增加得比產熱快($\dfrac{dQ_r}{dT} > \dfrac{dQ_g}{dT}$),系統會自己把擾動壓回去——這是穩定的。
寫成不等式,穩定穩態需滿足:
$$\frac{dQ_r}{dT} > \frac{dQ_g}{dT}$$
幾何上看就是「移熱直線比產熱曲線更陡」的那些交點才穩定。S 形曲線的低溫段與高溫段都比直線平緩(直線較陡)→ 穩定;中段最陡(陡過直線)→ 不穩定。這就解釋了開頭的怪現象:你升高冷卻水溫度 $T_c$,等於把整條移熱直線往右平移,低溫穩態與不穩定穩態會逐漸靠近、在某個臨界點相切後一起消失,系統別無選擇只能「跳」到僅存的高溫穩態——這就是點火(ignition)。反向降溫時,要降到另一個相切臨界點高溫穩態才消失、掉回低溫——這就是熄火(extinction)。兩個臨界溫度不相等,中間那段就是遲滯(hysteresis),正是你觀察到的「升上去和降回來走不同路」。
這套分析在工程上極其重要:反應器的點火條件決定了開車(startup)策略,熄火條件決定了它能容忍多大的擾動而不滅火。設計時若不小心把操作點放在不穩定穩態附近,反應器會在高低溫之間自發震盪,甚至演變成熱失控。入門篇那條 CSTR 代數式只給你「一個」解,而真實世界要求你回答「有幾個解、哪個穩定、怎麼到達它」。
機制二:真實流動的停留時間分布(RTD)
入門篇告訴你 PFR 裡每份流體待的時間都是 $\tau$、CSTR 是一個分布。進階篇要把這個「分布」變成可計算的數學工具,並用它預測非理想反應器的真實轉化率。
定義停留時間分布函數 $E(t)$:一份在 $t=0$ 進入反應器的流體,其中在 $t$ 到 $t+dt$ 之間離開的比例為 $E(t)\,dt$。它是一個機率密度函數,滿足歸一化:
$$\int_0^{\infty} E(t)\,dt = 1$$
平均停留時間就是它的一階矩,理想上應等於空間時間 $\tau$:
$$\bar{t} = \int_0^{\infty} t\,E(t)\,dt = \tau \quad(\text{理想無死區時})$$
兩種理想反應器的 $E(t)$ 是兩個極端:
- 理想 PFR:所有流體待的時間都精確等於 $\tau$,$E(t)$ 是一個位在 $t=\tau$ 的狄拉克脈衝 $\delta(t-\tau)$,變異數為零。
- 理想 CSTR:因為完全混合,剛進來的流體有相當機率立刻被沖出去,$E(t)$ 是指數衰減:
$$E(t) = \frac{1}{\tau}\,e^{-t/\tau}$$
真實反應器落在兩者之間。我們用脈衝示蹤實驗(pulse tracer)量它:在入口瞬間打入一團惰性示蹤劑,在出口連續量濃度 $C(t)$,則
$$E(t) = \frac{C(t)}{\displaystyle\int_0^{\infty} C(t)\,dt}$$
看一個例子:用 RTD 算非理想反應器的轉化率
RTD 最強的用途,是在不知道反應器內部流場細節的情況下,仍能估計轉化率。對一個一級反應(轉化率只依停留時間、與混合程度無關的特例),分離流(segregated flow)模型 給出:
$$\bar{X}_A = \int_0^{\infty} X_A(t)\,E(t)\,dt = \int_0^{\infty}\bigl[1 - e^{-kt}\bigr]E(t)\,dt$$
意思是:把每一「小包」流體按它各自待的時間 $t$ 算出轉化率 $1-e^{-kt}$,再用 $E(t)$ 加權平均。
來驗證它與入門篇結果一致。對理想 CSTR,代入 $E(t)=\frac{1}{\tau}e^{-t/\tau}$:
$$\bar{X}_A = \int_0^{\infty}\bigl(1 - e^{-kt}\bigr)\frac{1}{\tau}e^{-t/\tau}\,dt = 1 - \frac{1}{\tau}\int_0^{\infty}e^{-(k+1/\tau)t}\,dt = 1 - \frac{1/\tau}{k + 1/\tau} = \frac{k\tau}{1 + k\tau}$$
正好回到入門篇那條 CSTR 一級反應公式 $X_A = \dfrac{k\tau}{1+k\tau}$——RTD 框架把理想 CSTR 當成自己的特例復現了出來。但威力在於:給你一個從示蹤實驗量到的、長相歪七扭八的真實 $E(t)$,你照樣能套這條積分式估轉化率,而不必先解出反應器內複雜的流場。
一個必須點破的陷阱:分離流模型對一級反應給的答案是精確的,但對非一級反應,RTD 不足以唯一決定轉化率。原因是「分子何時相遇混合」這件事(micromixing)也會影響結果——同樣的 $E(t)$,可以對應「流體早早就在分子尺度混勻」或「各包流體各走各的、最後才混」兩種極端,兩者對二級或自催化反應算出的轉化率不同。RTD 只描述了巨觀混合(macromixing),真實反應器還需要微觀混合模型才能完整。這正是 RTD 理論從「工程估算工具」走向「研究級流體–反應耦合」的分水嶺。
機制三:觸媒顆粒內,擴散與反應誰先到?
入門篇提過異相觸媒有質傳阻力。進階篇我們把那顆觸媒顆粒剖開,從顆粒內部的微分方程把 Thiele 模數推出來,看清「擴散限制」到底是怎麼回事。
考慮反應物 $A$ 從顆粒外表面擴散進入一個多孔觸媒小球,邊擴散邊在孔壁上反應(一級)。對球內任一薄殼寫穩態質量平衡——「擴散進來的淨通量 = 在此殼層被反應消耗的量」——得到一條二階微分方程:
$$D_e\frac{1}{r^2}\frac{d}{dr}\!\left(r^2\frac{dC_A}{dr}\right) = k_v\, C_A$$
其中 $D_e$ 是有效擴散係數(effective diffusivity),$k_v$ 是以顆粒體積為基準的速率常數。把它無因次化,自然浮現一個無因次群——Thiele 模數(Thiele modulus) $\phi$:
$$\phi = R\sqrt{\frac{k_v}{D_e}}$$
它的物理意義一句話講完:$\phi$ 是「反應速率」與「擴散速率」的競賽指標。
- $\phi \ll 1$(反應慢、擴散快):反應物來得及鑽到顆粒最深處,整顆觸媒濃度幾乎均勻,全顆粒都在工作。
- $\phi \gg 1$(反應快、擴散慢):反應物才剛擴散進表面薄層就被反應掉了,顆粒中心根本「吃不到」反應物,大部分觸媒在睡覺。
我們用效率因子(effectiveness factor) $\eta$ 量化「整顆顆粒實際反應速率」相對於「假設全顆粒都處在表面濃度時的理想速率」的比值。解上面那條球座標方程,對一級反應可得到漂亮的封閉解:
$$\eta = \frac{3}{\phi}\left(\frac{1}{\tanh\phi} - \frac{1}{\phi}\right)$$
在 $\phi \gg 1$ 的強擴散限制區,這條式子簡化為 $\eta \approx 3/\phi$——意思是有效反應速率反而與 $\sqrt{k_v}$ 成正比(因為 $\phi\propto\sqrt{k_v}$,$\eta\propto 1/\sqrt{k_v}$,兩者相乘)。這帶來一個反直覺、卻在工程上致命的後果:當反應被擴散卡住時,你量到的「表觀活化能」只有真實活化能的一半。因為觀測速率 $\propto \eta k_v \propto \sqrt{k_v} \propto e^{-E_a/(2RT)}$,做阿瑞尼斯作圖時斜率減半。
動手試試:你量到的活化能是真的嗎?
假設你在實驗室量某觸媒反應的活化能,用入門篇教的 $\ln k$ 對 $1/T$ 作圖,得到 $E_a^{\text{obs}} = 40\ \text{kJ/mol}$。同事卻說文獻上這個本徵反應是 $80\ \text{kJ/mol}$。誰錯了?
很可能兩個都對——你量的是擴散限制區的表觀值(真值的一半),文獻量的是本徵動力學值。怎麼確認?把觸媒磨小。Thiele 模數 $\phi \propto R$,顆粒半徑減半,$\phi$ 減半。若你把顆粒磨碎後反應速率明顯變快、且重新作圖斜率變陡(活化能回到 $80$),就證明原本被擴散卡住了。這個「磨小顆粒測速率」的判斷法(配合 Weisz–Prater 準則 $\phi^2\eta = \dfrac{(-r_{A,\text{obs}})R^2}{D_e C_{As}}$ 是否 $\ll 1$)是異相反應工程的標準診斷流程。設計觸媒時刻意做小顆粒或薄殼觸媒(egg-shell catalyst)來逼近 $\eta \to 1$,正是這套理論的直接應用。
把三個機制接回優化學
入門篇把反應器設計寫成一個帶守恆約束的最佳化問題。進階篇的三個機制,恰好揭示了那個最佳化問題裡最棘手的三類約束:
$$\max_{T,\,\tau,\,R_p,\,\text{network}} \; \big[\,\text{利潤} = p_B\,\dot{n}_B - c_{\text{操作}} - c_{\text{設備}}\,\big]$$
- 多重穩態讓可行域變得非凸(non-convex)且不連續:操作點若落在點火–熄火遲滯區內,目標函數會跳變,梯度型優化器會被「卡」在錯誤的局部解,必須用全域優化或刻意避開不穩定分支。
- RTD 把「反應器內部混合程度」這個原本被理想化掉的東西,變成設計變數——串聯幾個 CSTR、用多大的軸向擴散,都會改變 $E(t)$ 進而改變選擇性,這是反應器網絡綜合(reactor network synthesis) 的核心,現代做法用 可達性網(attainable region) 在濃度空間中直接畫出所有可能達到的產物分布,找出最佳反應器組合。
- Thiele 模數把顆粒半徑 $R_p$、孔隙率這類觸媒微結構參數也拉進決策變數:太大顆粒壓降低但 $\eta$ 差,太小顆粒 $\eta$ 好但床層壓降暴增——又一個 trade-off。
於是程序模擬軟體裡的「一個反應器模組」,背後其實是同時在解非線性穩態多重解、RTD 卷積、與顆粒內擴散方程。優化學負責在這個崎嶇的地形上找路,而反應工程負責把地形本身畫對——模型錯了,再強的優化器也只會精準地給你一個錯誤答案。
重點回顧
- 多重穩態源於非線性:放熱 CSTR 的產熱對溫度呈 S 形、移熱呈直線,可交三點。穩定判據是「移熱比產熱更陡」($dQ_r/dT > dQ_g/dT$);升降冷卻溫度造成點火–熄火遲滯,是入門代數式看不到的真實行為。
- RTD 把混合量化:$E(t)$ 是停留時間的機率密度,理想 PFR 是脈衝、理想 CSTR 是指數衰減。分離流模型 $\bar{X}_A=\int(1-e^{-kt})E(t)\,dt$ 對一級反應精確,且能復現入門 CSTR 公式。
- RTD 不是萬能:它只描述巨觀混合,對非一級反應還需微觀混合(micromixing)模型才能定轉化率。
- Thiele 模數是擴散 vs 反應的競賽:$\phi=R\sqrt{k_v/D_e}$,大 $\phi$ 時顆粒中心吃不到反應物,效率因子 $\eta\approx 3/\phi$,表觀活化能掉到真值一半。
- 磨小顆粒可診斷擴散限制:$\phi\propto R$,縮小顆粒讓 $\eta\to 1$;Weisz–Prater 準則可用觀測量直接判斷是否落在擴散區。
- 進階機制是優化的硬約束:多重穩態造成非凸、RTD 牽動網絡綜合、Thiele 模數牽動顆粒設計,三者讓反應器最佳化遠比理想手算複雜。
深入探討(研究所視角)
把三個機制再往前推,會通向幾個當代反應工程的活躍研究前沿:
1. 動態行為與分岔分析(dynamic & bifurcation analysis)。 本文只談穩態的多重解,但反應器是動態系統。用連續性方法(continuation)追蹤穩態隨參數的變化,會畫出分岔圖(bifurcation diagram):點火–熄火對應鞍結分岔(saddle-node bifurcation),而某些參數下系統會出現 Hopf 分岔 進入持續的自激震盪(自發的溫度–濃度極限環)。理解這些非線性動力學對反應器的安全操作與控制器設計至關重要,是化工與應用數學的交界。
2. 計算流體力學耦合反應(CFD-reaction coupling)。 RTD 給的是「黑箱」式的巨觀混合資訊,現代研究直接用 CFD 解出反應器內部的速度場、濃度場與溫度場,把 Navier–Stokes 方程與反應源項耦合求解。這讓我們能設計攪拌槳幾何、消除死區、優化進料口位置,把「非理想」從事後修正變成事前設計。對快速反應與強放熱系統,micromixing 模型(如 IEM、PDF 方法)更是不可或缺。
3. 多尺度建模(multiscale modeling)。 觸媒反應橫跨從原子尺度(活性位點上的吸附–反應,用 DFT 與微觀動力學 microkinetics 算)、到顆粒尺度(孔內擴散–反應,本文的 Thiele 分析)、到反應器尺度(床層流動與熱傳)。如何把這三個尺度的模型一致地接起來,讓分子層級的觸媒設計能預測整座反應器的表現,是當代反應工程結合計算化學的核心挑戰。
4. 操作優化與機器學習(real-time optimization & ML surrogates)。 因為完整模型(含多重穩態、CFD、多尺度)求解極慢,難以用於即時控制,研究者訓練代理模型(surrogate model) 或物理資訊神經網路(PINN)來逼近反應器行為,搭配模型預測控制(MPC)做即時最佳化。挑戰在於——代理模型必須忠實重現多重穩態與遲滯這類強非線性結構,否則控制器會在點火–熄火邊界做出危險決策。這把資料驅動方法、非線性動力學與傳統反應工程縫在一起,正是這個領域最前沿的縫合處。
把這四條線收攏,你會看到進階反應工程的真正主題:入門篇的理想反應器是一個「點」,進階篇研究的是這個點周圍的整片地形——它如何隨溫度分岔、隨流場扭曲、隨尺度變形。化學工程把分子尺度的化學放大成工業生產,而能否安全、高效、永續地放大,取決於我們對這片非線性地形理解得有多深。