
一片平板電極的電流,為什麼從來不會「均勻」?
從電流分佈、極限電流密度到多孔電極與反應器放大,看電化學如何成為一門可設計、可優化的反應器工程。
一片平板電極的電流,為什麼從來不會「均勻」?
你已經知道,一個電解槽的操作電壓是 $E_{\text{cell}} = E_{\text{eq}} + |\eta_{\text{act}}| + |\eta_{\text{conc}}| + iR_\Omega$,也知道法拉第定律能把電量換算成產量。但這些式子背後藏著一個入門篇沒說破的假設:它們都假設整片電極上的電流密度 $i$ 是一個單一的數字。
真實世界裡,這個假設幾乎總是錯的。把一片 1 公尺見方的平板電極浸進電解液通電,靠近進電端、靠近邊緣、靠近對極的地方,電流密度可以差到好幾倍。某些區域被「操爆」——過電位過高、副反應失控、電極被燒蝕;另一些區域卻幾乎沒在工作,白白佔了昂貴的觸媒面積。一座電解廠的效率、壽命與成本,往往不取決於平均電流密度,而取決於電流分佈(current distribution)有多不均。
這就是電化學工程從「一顆理想電池」走向「一座真實反應器」時,必須面對的核心難題。這篇進階文章,談的就是如何把電化學放大成可設計的反應器——當電極不再是一個點,而是一個有體積、有流場、有電位梯度的空間時,我們該怎麼算、怎麼設計、怎麼優化。
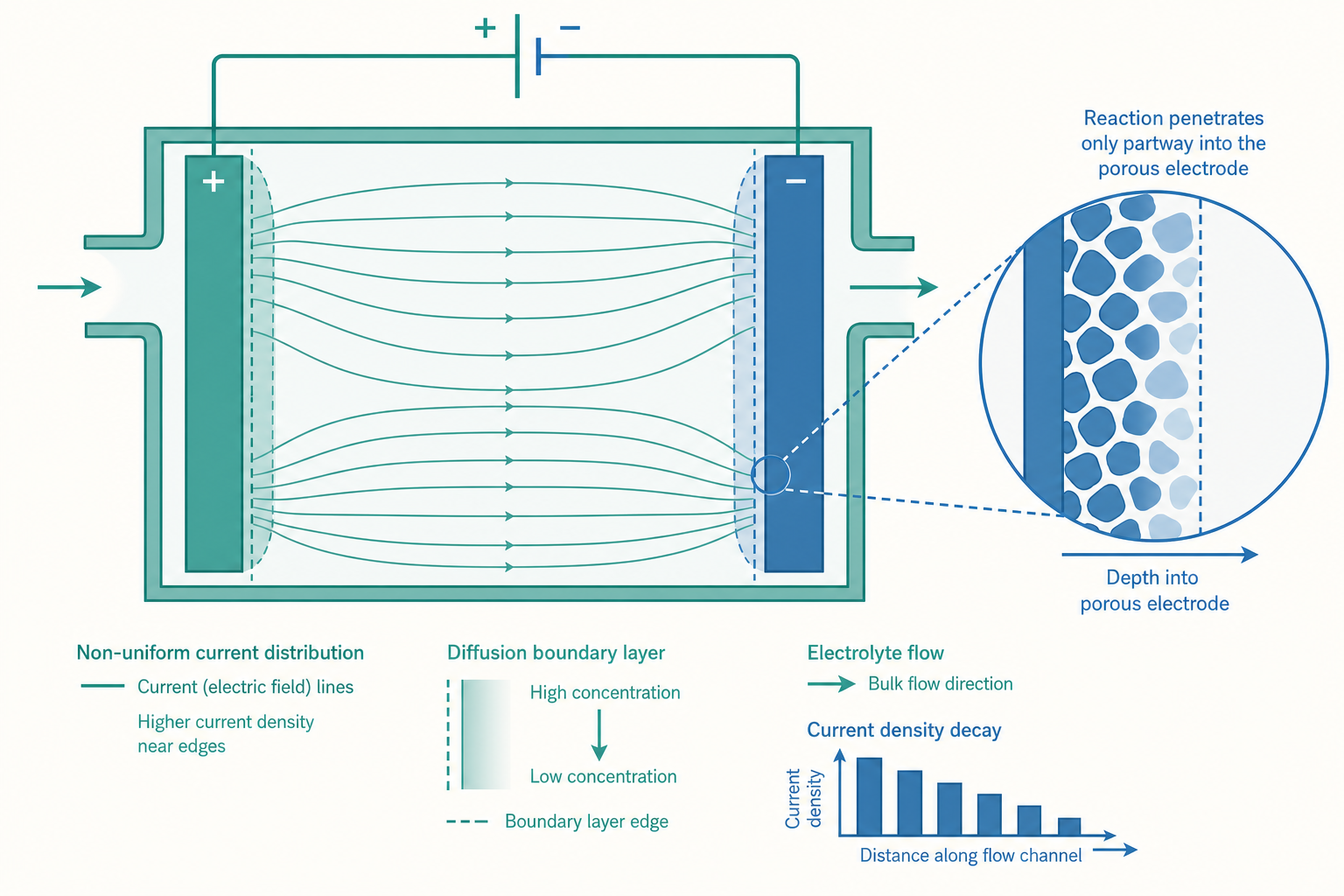
三種電流分佈:把過電位拆開來看空間
要理解電流為什麼不均,得先承認電解槽裡同時有「兩個電阻網路」在競爭:電解液的離子電阻,與電極反應本身的「反應電阻」(過電位對電流的阻礙)。電流會挑阻力小的路走,這兩個電阻誰主導,就決定了分佈的型態。電化學工程把它分成三個層次:
一次電流分佈(primary current distribution)。 假設電極反應快到完全不需要過電位($\eta \approx 0$),整個系統只剩電解液的歐姆電阻在主導。這時電流純粹由幾何決定——哪裡離對極近、電位梯度大,電流就往哪裡擠。結果是最不均勻的分佈:平板邊緣與尖角的電流密度趨於發散,這也是為什麼電鍍件的稜角總是最先鍍厚、甚至燒焦。一次分佈由拉普拉斯方程(Laplace's equation)支配:
$$ \nabla^2 \phi = 0 $$
其中 $\phi$ 是電解液中的電位。它的解只跟槽體形狀與電極位置有關,與電流大小無關。
二次電流分佈(secondary current distribution)。 現在把活化過電位放回來。電極反應需要過電位才能跑,而過電位高的地方反應「比較吃力」,於是電流會被推離高電流區、攤向低電流區——過電位扮演了「自動均勻化」的角色。一次分佈裡電流密度本來要發散的尖角,因為一旦電流大、過電位就跟著大、反而把電流頂回去,分佈因此被撫平。決定二次分佈均勻程度的,是一個無因次群——華格納數(Wagner number):
$$ \mathrm{Wa} = \frac{\kappa}{L} \cdot \frac{\partial \eta}{\partial i} = \frac{\kappa \, (\partial \eta / \partial i)}{L} $$
其中 $\kappa$ 是電解液導電度($\mathrm{S/m}$),$L$ 是電極特徵長度(m),$\partial\eta/\partial i$ 是極化曲線在操作點的斜率(過電位對電流密度的敏感度)。它的物理意義很乾淨:
- $\mathrm{Wa} \ll 1$:歐姆電阻主導,反應電阻可忽略 → 趨近一次分佈,很不均勻。
- $\mathrm{Wa} \gg 1$:反應電阻主導,過電位把電流攤平 → 很均勻。
工程師想讓電鍍層厚薄一致、想讓電解槽各處均勻老化,本質上就是想方設法提高 Wagner 數:用更高導電度的電解液、縮小電極尺寸、選擇極化曲線較「軟」(斜率大)的反應條件。
三次電流分佈(tertiary current distribution)。 再把濃度過電位、也就是質傳的影響加進來,分佈就進入最完整、也最難算的層次。此時電極表面反應物的局部濃度會隨電流變化,反過來改變局部的反應速率,三者(歐姆、活化、濃度)彼此耦合。這也把我們帶到下一個關鍵概念。
極限電流密度:質傳設下的天花板
入門篇把濃度過電位描述成「反應太快、反應物來不及補充」。進階一點看,這件事有一個硬上限:無論你把電壓加到多高,電極所能維持的電流密度,被反應物從液相主體擴散到電極表面的速率「卡死」。這個上限叫極限電流密度(limiting current density)$i_L$。
把電極表面想成一層薄薄的擴散邊界層(diffusion boundary layer),厚度 $\delta$。穩態下,反應物穿過這層的擴散通量決定了最大可能電流。由費克定律(Fick's law):
$$ i_L = \frac{n F D \, C_b}{\delta} = n F \, k_m \, C_b $$
其中 $D$ 是反應物擴散係數($\mathrm{m^2/s}$),$C_b$ 是液相主體濃度($\mathrm{mol/m^3}$),$\delta$ 是邊界層厚度,$k_m = D/\delta$ 是質傳係數(mass transfer coefficient)。當操作電流逼近 $i_L$ 時,電極表面濃度趨近於零,濃度過電位急遽飆升,電壓暴衝——再加電壓也擠不出更多電流,只會把多餘能量全變成熱與副反應。
這條式子是電化學反應器設計的鐵律,而它的關鍵在 $k_m$:質傳係數可以被工程手段操控。 加快流速、設計湍流促進器(turbulence promoter)、用旋轉電極、改用多孔結構增加比表面積——全都是在壓薄 $\delta$、放大 $k_m$,從而把 $i_L$ 這片天花板往上頂。$k_m$ 通常由無因次關聯式估算:
$$ \mathrm{Sh} = a \, \mathrm{Re}^{b} \, \mathrm{Sc}^{c} $$
其中 $\mathrm{Sh} = k_m L / D$ 是雪伍德數(Sherwood number)、$\mathrm{Re}$ 是雷諾數、$\mathrm{Sc} = \nu/D$ 是施密特數,$a,b,c$ 為與流道幾何相關的經驗常數。看到這裡你應該會心一笑:這正是質量傳遞那一章的關聯式,原封不動搬進電化學。 電化學反應器,骨子裡就是一個「帶電場的質傳設備」。
多孔電極:用第三個維度換面積
如果反應器的瓶頸是面積與質傳,最聰明的解法之一是:別用平板,用多孔電極(porous electrode)。 燃料電池的氣體擴散層、液流電池的碳氈、電容去離子的活性碳——這些電極內部佈滿孔隙,把「電化學反應的舞台」從一個二維表面,拓展成一個三維體積。同樣的外觀尺寸,比表面積(specific surface area, $a$,單位 $\mathrm{m^2/m^3}$)可以放大成千上萬倍。
但天下沒有白吃的午餐。多孔電極內部,電子要在固體骨架裡傳導、離子要在孔隙電解液裡傳導,兩條路徑都有電阻,於是反應不會均勻發生在整個厚度上。靠近電解液側的反應劇烈、深處可能幾乎沒反應——電極「深度」沒被充分利用。描述這個現象的,是 Newman 多孔電極理論裡的耦合方程組。在一維、忽略濃度變化的簡化下,固相電位 $\phi_s$ 與液相電位 $\phi_l$ 滿足:
$$ \sigma \frac{d^2 \phi_s}{dx^2} = a \, i_{loc}(\eta), \qquad \kappa \frac{d^2 \phi_l}{dx^2} = -a \, i_{loc}(\eta), \qquad \eta = \phi_s - \phi_l - E_{eq} $$
其中 $\sigma$ 是固相導電度、$\kappa$ 是液相導電度、$i_{loc}$ 是局部反應電流密度(由 Butler–Volmer 決定)。這組式子的解,會給出一個沿厚度衰減的反應分佈,特徵衰減長度(penetration depth)約為:
$$ \ell \sim \sqrt{\frac{1}{a \, i_0 \, (nF/RT)} \cdot \frac{\sigma \kappa}{\sigma + \kappa}} $$
工程上的設計準則由此而生:電極厚度若遠大於 $\ell$,加厚只是浪費材料;若遠小於 $\ell$,又沒充分利用三維優勢。 最佳厚度落在與 $\ell$ 同數量級處。這也解釋了為什麼鉑這類昂貴觸媒,要做成奈米顆粒分散在多孔載體上——既要最大化 $a$,又要讓反應均勻滲透整個厚度,把每一克貴金屬的價值榨乾。
看一個例子:水電解槽的電流密度該設多高?
假設你要設計一座鹼性水電解產氫槽,陰極反應為 $\mathrm{2H_2O + 2e^- \rightarrow H_2 + 2OH^-}$。已知操作參數:電解液導電度 $\kappa = 60\ \mathrm{S/m}$,兩極間距 $L = 3\ \mathrm{mm} = 0.003\ \mathrm{m}$,極化曲線在操作點的斜率 $\partial\eta/\partial i \approx 1.5 \times 10^{-4}\ \mathrm{V\cdot m^2/A}$(即每增加 $1\ \mathrm{A/m^2}$,過電位增加約 $0.15\ \mathrm{mV}$)。我們來評估電流分佈與歐姆損失。
第一步:估算 Wagner 數,判斷均勻度。
$$ \mathrm{Wa} = \frac{\kappa \,(\partial\eta/\partial i)}{L} = \frac{60 \times 1.5\times10^{-4}}{0.003} = \frac{9\times10^{-3}}{0.003} = 3.0 $$
$\mathrm{Wa} = 3 > 1$,代表反應電阻略佔上風,電流分佈相當均勻——這是好消息,整片電極會被均衡使用,不易出現局部燒蝕。
第二步:估算歐姆壓降,看能耗代價。 取操作電流密度 $i = 4{,}000\ \mathrm{A/m^2}$(商用電解槽典型值),電解液歐姆壓降為:
$$ \Delta V_\Omega = i \cdot \frac{L}{\kappa} = 4{,}000 \times \frac{0.003}{60} = 0.20\ \mathrm{V} $$
第三步:連結優化學——電流密度的權衡。 假設此槽在 $i=4{,}000\ \mathrm{A/m^2}$ 下總槽電壓約 $E_{\text{cell}} = 1.9\ \mathrm{V}$(含 $E_{eq}\approx1.23\ \mathrm{V}$、活化過電位、與上面的歐姆項)。每生產 $1\ \mathrm{mol}$ 氫氣需 $2$ 莫耳電子,理論電量 $Q = 2F = 192{,}970\ \mathrm{C}$,對應電能:
$$ W = E_{\text{cell}} \cdot Q = 1.9 \times 192{,}970 \approx 3.67 \times 10^{5}\ \mathrm{J/mol\ H_2} $$
換算成每公斤氫($1\ \mathrm{kg\ H_2} = 500\ \mathrm{mol}$):
$$ W = 3.67\times10^5 \times 500 = 1.83\times10^8\ \mathrm{J/kg} \approx 51\ \mathrm{kWh/kg\ H_2} $$
這個數字直指綠氫的命門:理論最低能耗約 $39\ \mathrm{kWh/kg}$(對應 $1.23\ \mathrm{V}$),現實中那多出來的 $\sim12\ \mathrm{kWh/kg}$,整個就是過電位與歐姆損失。若把電流密度從 $4{,}000$ 拉高到 $8{,}000\ \mathrm{A/m^2}$,產量翻倍、設備變小(CAPEX 降),但歐姆壓降也翻倍到 $0.40\ \mathrm{V}$、活化過電位上升,槽電壓升到約 $2.1\ \mathrm{V}$,能耗升到約 $56\ \mathrm{kWh/kg}$(OPEX 升)。最佳電流密度,就是讓「攤提的設備成本 + 電力成本」總和最小的那一點——這是一道標準的程序優化問題,骨架與入門篇那道電鍍題完全相同,只是把「時間」換成了「電流密度」這個設計變數。
把單槽變成工廠:反應器構型與放大
實驗室裡一個燒杯兩根電極,到工廠裡是上百片電極堆疊的電解槽列。放大時,電化學反應器的構型選擇,和一般化學反應器一樣有「混合程度」的光譜:
- 平行板槽(parallel-plate cell) 接近柱塞流反應器(PFR):電解液沿流道流過,反應物濃度沿程下降,轉化率高,但出口端因濃度低、$i_L$ 也低,容易遇到質傳瓶頸。
- 連續攪拌槽式(CSTR-like)構型:槽內充分混合、濃度均勻,操作穩定好控制,但出口濃度等於槽內濃度,轉化率受限。
兩者的物料平衡,可以直接套用反應工程的框架。以一個單程通過的平行板電解槽為例,反應物的穩態質量平衡為:
$$ Q_v \frac{dC}{dz} = -\frac{i(z) \, a_e}{nF} $$
其中 $Q_v$ 是體積流率($\mathrm{m^3/s}$)、$z$ 是沿流道座標、$a_e$ 是單位長度的電極面積。若反應落在質傳控制區($i = i_L = nF k_m C$),代入整理可得濃度沿程指數衰減:
$$ \frac{C_{out}}{C_{in}} = \exp\!\left(-\frac{k_m \, A_e}{Q_v}\right) $$
這個結果美在它的形式——它和「填充床反應器轉化率隨停留時間指數衰減」是同一個數學長相。 指數裡的 $k_m A_e / Q_v$ 扮演了電化學版的 Damköhler 數,比較「質傳能力」與「對流帶走的速率」。電化學反應器設計,因此可以完全嫁接到你已經熟悉的反應工程與輸送現象工具箱裡:要提高轉化率,就增大 $k_m$(強化質傳)、增大 $A_e$(多孔電極或更多板)、或降低 $Q_v$(拉長停留時間)——三條路,每一條都對應一筆成本,每一個權衡都是優化學的題目。
重點回顧
- 電流密度從不均勻。 電化學反應器的效率與壽命,常取決於電流分佈而非平均值;一次分佈(純歐姆,最不均)、二次分佈(加活化過電位,被撫平)、三次分佈(再加質傳,最完整)是三個遞進的分析層次。
- Wagner 數 $\mathrm{Wa} = \kappa(\partial\eta/\partial i)/L$ 量化分佈均勻度:$\mathrm{Wa}\gg1$ 均勻、$\mathrm{Wa}\ll1$ 不均。提高導電度、縮小電極、選軟極化曲線都能讓電流更均勻。
- 極限電流密度 $i_L = nFk_mC_b$ 是質傳設下的硬天花板。它的關鍵 $k_m$ 由 $\mathrm{Sh}=a\,\mathrm{Re}^b\mathrm{Sc}^c$ 估算——電化學反應器本質上是「帶電場的質傳設備」。
- 多孔電極用第三維度換取巨大比表面積,但反應沿厚度衰減;最佳厚度與穿透深度 $\ell$ 同數量級,這是貴金屬觸媒奈米化、分散化的根本原因。
- 放大設計可直接嫁接反應工程:平行板槽近似 PFR,質傳控制下濃度沿程指數衰減,$k_mA_e/Q_v$ 是電化學版 Damköhler 數。最佳電流密度由 CAPEX 與 OPEX 的權衡決定。
深入探討(研究所視角)
從進階走向研究前沿,電化學反應器工程的難點在於多物理場的強耦合與跨尺度建模。以下幾條主線值得深究:
一、完整的耦合場求解。 真實電解槽要同時解四組守恆:離子輸送(Nernst–Planck,含擴散/遷移/對流)、電荷守恆($\nabla\cdot\mathbf{i}=0$)、動量守恆(Navier–Stokes,給出流場 $\mathbf{v}$)與能量守恆(焦耳熱導致溫度場、進而改變 $\kappa$、$D$、$i_0$)。氣體析出型反應(產氫、產氧、產氯)更棘手——氣泡會遮蔽電極面積、降低有效導電度,需引入兩相流與空泡率(void fraction)模型。這類問題幾乎只能靠有限元素/有限體積數值模擬(如 COMSOL、自寫求解器)處理,是計算電化學(computational electrochemistry)的核心戰場。
二、暫態與循環操作。 入門與本文多半談穩態,但電池充放電、再生能源驅動的電解槽(風光出力波動)本質上是暫態。鋰電池的固相擴散要解球座標下的擴散方程(Doyle–Fuller–Newman, DFN 模型),SEI 膜生長、鋰枝晶(dendrite)成核則涉及相場(phase-field)建模。電解槽在波動電力下頻繁啟停,會加速觸媒劣化與膜降解——如何在動態工況下兼顧效率與壽命,是綠氫產業化的關鍵研究題。
三、反應器級的多目標優化。 把本文的物理串成最適化問題:以電流密度分佈、流道幾何、電極厚度、流速為設計變數,目標函數是「單位產品的均化總成本(levelized cost)」,限制式包括目標產量、最高局部過電位(保護電極)、最低電流效率、與溫升上限。這是一個受偏微分方程約束的最佳化(PDE-constrained optimization),需用伴隨法(adjoint method)求梯度,是程序系統工程與計算數學的交會點,也是把「優化學」從一道代數題提升為一個設計框架的所在。
四、前沿構型與系統整合。 當代研究正把電化學反應器推向新形態:零間隙(zero-gap)膜電極組合把 $L\to0$ 以消滅歐姆損失、陰離子交換膜(AEM)電解避開貴金屬、$\mathrm{CO_2}$ 氣體擴散電極把氣相反應物直送三相界面、液流電池解耦「功率」與「容量」以服務電網級儲能。這些題目共同把電化學反應器嵌入更大的能源與化工系統——與再生能源、碳捕集、氫經濟耦合。支撐它們向上放大的,始終是本文這套「電流分佈 × 質傳極限 × 反應器物料平衡 × 成本優化」的工程骨架。